
Aqui reunimos algumas das dúvidas mais comuns a respeito do estudo da genômica viral. Esta página foi criada durante a pandemia do coronavírus SARS-CoV-2, e agora tem foco na vigilância genômica também em outros contextos. Outros tópicos importantes que podem gerar dúvida na população também estão contemplados. O conteúdo desta página está em constante revisão e novas perguntas e respostas serão adicionados ao longo do tempo.
PERGUNTAS FREQUENTES
A Rede Genômica Fiocruz é um conjunto de grupos de pesquisa de diversas Unidades da Fundação Oswaldo Cruz presentes em todas as regiões do Brasil, trabalhando coordenadamente para o estudo da genética e genômica de microrganismos relevantes para a saúde humana.
A Rede Genômica foi criada para coordenar esforços para o sequenciamento e estudo do genoma do SARS-CoV-2, coronavírus causador da COVID-19, como parte do esforço global para acompanhar a dispersão do vírus por diferentes regiões do mundo. O foco desta atuação está na identificação e rastreio de suas mutações genéticas e no surgimento de novas linhagens. Atualmente, a Rede Genômica expandiu seu escopo para a vigilância e estudo do genoma de outros patógenos, como os vírus Influenza e Dengue.
Para mais informações sobre a Rede e as Instituições parceiras, acesse a página “A Rede” do nosso site.
O sequenciamento genômico de alto desempenho realizado em amostras do novo coronavírus (SARS-CoV-2), além de permitir o monitoramento da circulação de diferentes linhagens do vírus, a detecção de padrões na emergência de novas variantes e no seu espalhamento em relação a outras linhagens e a identificação de variantes ainda não descritas, permite também o estudo de mutações relevantes do vírus.
Compreender como o vírus causador da Covid-19 se espalha entre diferentes regiões, e monitorar a prevalência de suas diferentes linhagens genéticas, foi um processo de muita relevância para o planejamento estratégico do combate à pandemia. Esses dados são de grande importância para a elaboração de testes de diagnóstico específicos para novas variantes, atualização de vacinas e delineamento de medidas de saúde pública para a contenção da doença. Estas ações de monitoramento, vigilância e delineamento de medidas de saúde pública continuam relevantes mesmo após o final do status pandêmico do vírus, pois permitem continuamente estudar as linhagens que circulam na população.
O estudo de alterações genéticas tais como aquelas na estrutura da proteína Spike — através da qual o vírus entra nas células hospedeiras — é importante também para o entendimento dos riscos associados à circulação destas variantes mutantes. Este conhecimento permite a identificação de mutações que podem servir como “sinais de alerta”, ao surgirem de forma independente em locais distintos.
A Rede Genômica resulta do trabalho coordenado de laboratórios e grupos de pesquisa em 12 unidades da Fiocruz, que já realizavam tradicionalmente estudos com a genômica dos mais diversos organismos de interesse para a saúde humana antes da pandemia de Covid-19. Desta forma, o sequenciamento e análise do material genético de organismos causadores de doenças como a Leishmania, viroses respiratórias como a influenza (gripe) e o vírus sincicial respiratório, e outras doenças como dengue, sarampo e esquistossomose já eram parte da rotina dos laboratórios que constituem a rede antes da emergência de saúde coletiva em 2020.
No âmbito do estudo do SARS-CoV-2, do Influenza e do Dengue, a Rede Genômica estuda as sequências genéticas de amostras recolhidas em todo o Brasil, abastecendo o banco de dados da iniciativa internacional GISAID e identificando as linhagens virais que circulam nas diferentes regiões do país.
Os grupos de pesquisa que compõem a Rede Genômica trabalham há anos com o estudo dos mais diversos aspectos do genoma de organismos que causam doenças infecciosas. Graças à pesquisa de laboratórios que hoje fazem parte da rede, entendemos melhor os mecanismos de patogênese — ou seja, os mecanismos através dos quais um organismo ou vírus causa um quadro clínico — o espalhamento, a evolução e o genoma dos agentes causadores de doenças relevantes para a população brasileira.
Estes incluem estudos focados em arboviroses como a dengue e a zika, além de parasitoses tropicais como Leishmaniose e a doença de Chagas. Mapear locais de ocorrência destas doenças, em conjunção com a análise genômica de amostras em cada localidade, ajuda na detecção de zonas de maior risco, na formulação de estratégias de controle e prevenção, e na compreensão da evolução dos agentes causadores. O estudo do genoma destes patógenos também é importante para uma compreensão mais detalhada dos mecanismos de cada doença, que abre portas para o desenvolvimento de abordagens terapêuticas e/ou profiláticas (como vacinas) para combatê-las e devolver a saúde a pacientes acometidos e a suas famílias.
De 2020 a 2023 estas pesquisas ficaram concentradas no estudo do coronavírus SARS-CoV-2, e na compreensão de múltiplos aspectos da pandemia causada por este patógeno relativamente novo. Após o final do status pandêmico do SARS-CoV-2, os esforços da Rede Genômica se expandiram para outros agravos de relevância para o cenário brasileiro, como arboviroses (em especial a Dengue) e o vírus Influenza, além de manter a vigilância do SARS-CoV-2.
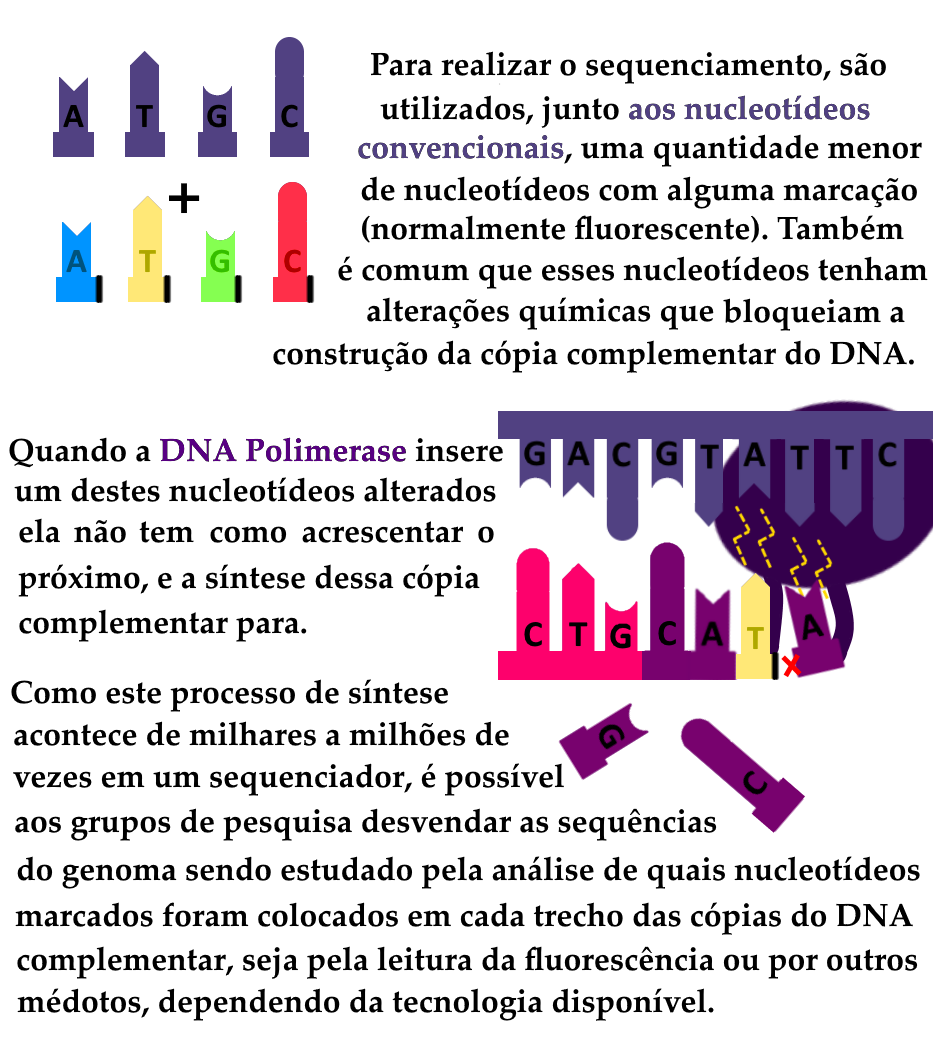
O sequenciamento genético é uma técnica baseada em reações químicas que já acontecem naturalmente nas células quando o material genético é replicado. Associando as enzimas utilizadas nessa duplicação a uma tecnologia envolvendo fluorescência, as técnicas de sequenciamento permitem saber parte ou toda a informação presente no código genético de um organismo. Ele permite, portanto, identificar genes, predizer seus produtos (proteínas, RNAs de interferência, ribozimas, etc) e comparar sequências de organismos diferentes.
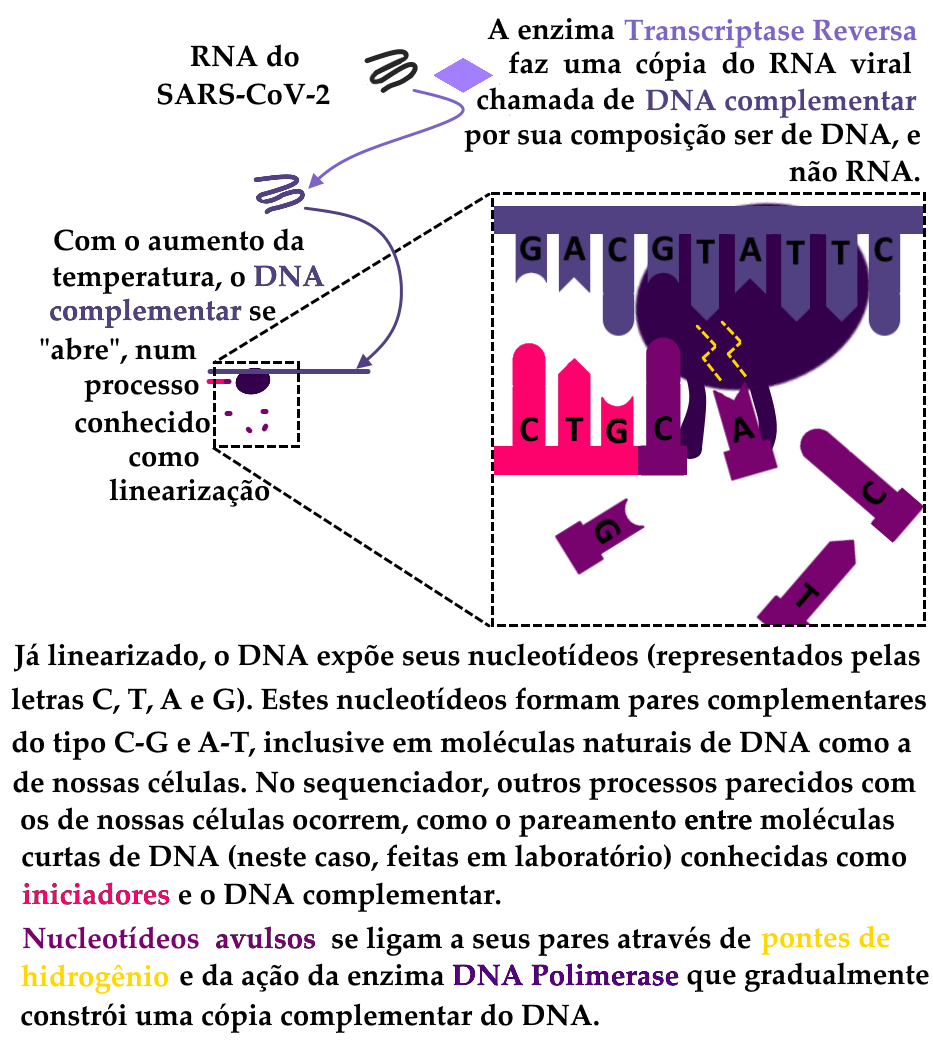
A partir desta comparação, entre sequências de amostras mais antigas do SARS-CoV-2 e sequências de amostras mais recentes, é possível identificar os pontos em comum e os pontos de divergência entre o genoma das mesmas. Essa análise permite traçar linhas evolutivas e analisar o parentesco entre variantes. Desta forma, amostras de vírus que possuam uma mesma origem, e se diferenciaram a partir de um mesmo ancestral, são classificadas em uma mesma linhagem, e pode-se entender também como uma linhagem dá origem a outras. Esta mesma abordagem pode ser utilizada para outros vírus e organismos.
Em qualquer evento epidêmico de larga escala, especialmente aqueles caracterizados por um rápido espalhamento do agente etiológico, o monitoramento de casos se faz importante para que se possa acompanhar a dinâmica da epidemia. O perfil genético do vírus traz informações sobre seu espalhamento sob os pontos de vista geográfico e temporal, revelando suas principais rotas de transmissão. Em outras palavras, o genoma é a identidade do patógeno e revela por onde e quando ele se disseminou.
Após um ano do surgimento do SARS-CoV-2, o sequenciamento genético assume papel ainda mais central no combate à pandemia devido ao constante surgimento de novas variantes do vírus, classificadas naquele momento como “variantes de interesse” (aquelas que devem ser monitoradas atentamente, mas que que apresentam menor probabilidade de estarem associadas a quadros mais graves ou maior infectividade) e “variantes de preocupação” (aquelas que possivelmente trazem implicações substanciais para a saúde coletiva). O estudo das mutações permite ainda um maior entendimento dos mecanismos de patogenicidade, ou seja, os genes associados ao surgimento da doença, à sua gravidade ou capacidade de infectar novos hospedeiros.
O monitoramento de arboviroses endêmicas como a dengue também tem uma grande importância, uma vez que é possível acompanhar eventos como a recente reintrodução de um sorotipo do vírus da Dengue que não circulava no Brasil há aproximadamente 30 anos. A detecção de eventos como estes é importante para embasar a formulação de políticas públicas de vigilância e enfrentamento destes agravos.
A anotação de genomas consiste na análise dos dados gerados após o sequenciamento, ou seja, no estudo minucioso das sequências de informação genética, auxiliado pelo computador. O processo de anotação envolve a identificação de estruturas que marcam regiões do genoma, como o início e o final de genes, as sequências envolvidas na regulação da expressão (ativação ou desativação) de genes, e, eventualmente, regiões que não são expressas. A anotação é, portanto, a interpretação e organização da informação genética. Ela pode auxiliar, por exemplo, na comparação de versões diferentes de um mesmo gene, presentes em amostras de linhagens diferentes, e entender quais são as alterações nestes genes (e nas sequências próximas, que podem influenciar sua expressão). Desta maneira, ao desvendar o sentido das sequências, a anotação é uma etapa imprescindível do estudo do genoma do novo coronavírus, e na classificação das amostras em linhagens.
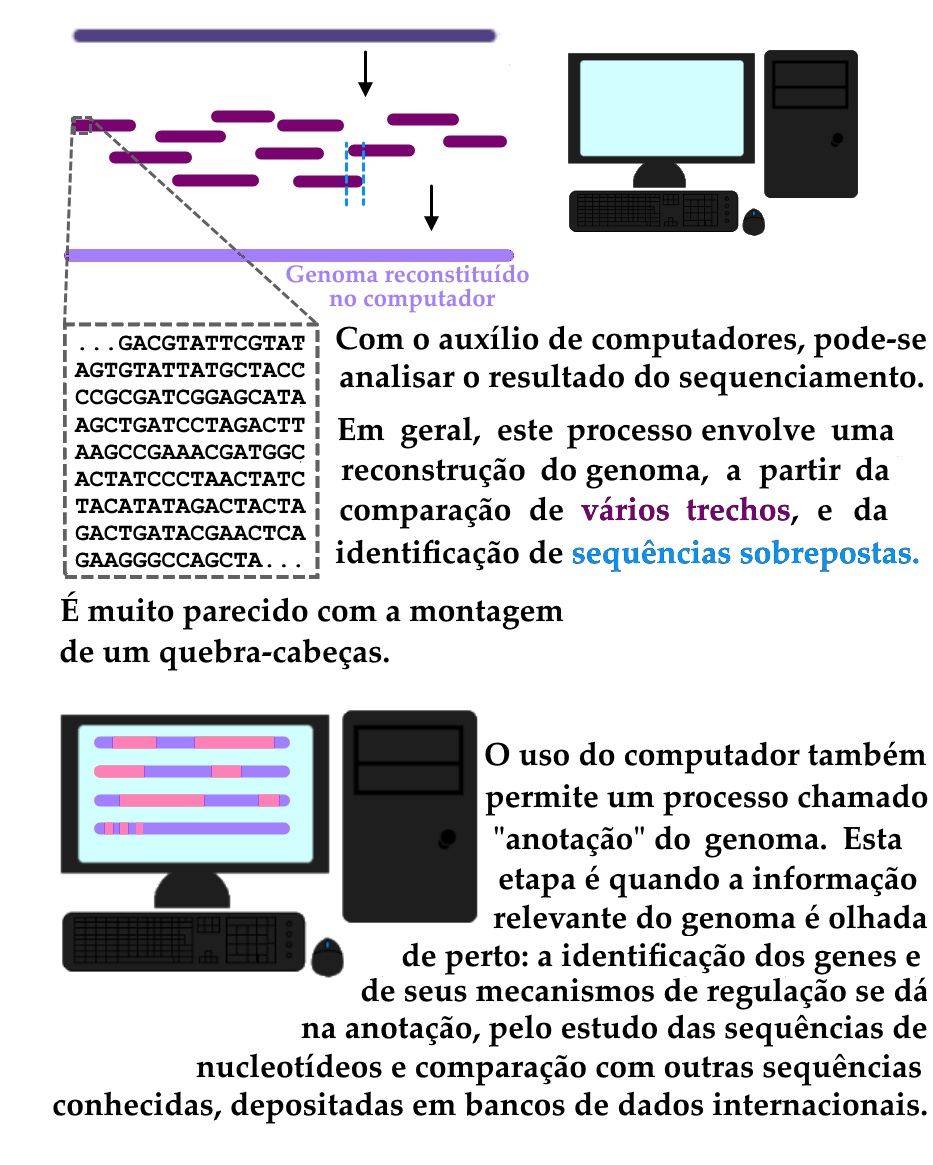
Compreender o genoma de um agente infeccioso — seja um vírus, uma bactéria, fungo ou parasito — permite uma série de abordagens para o combate à doença por ele causada. No que diz respeito ao desenvolvimento de tratamentos, a análise de sequências genéticas trazem informações sobre a fisiopatologia do vírus, identificando possíveis alvos para intervenção terapêutica.
Adicionalmente, o estudo de genes envolvidos na patogenia pode auxiliar no desenvolvimento de novas terapias por meio de outra abordagem: a modelagem molecular tridimensional. Através de ferramentas de bioinformática, é possível construir modelos de proteínas importantes, como a Spike do novo coronavírus, envolvida na invasão das células hospedeiras. Com base no estudo das sequências genéticas responsáveis pela expressão desta proteína, é possível não apenas predizer sua estrutura e desenvolver moléculas que bloqueiem sua ligação aos receptores presentes nas células humanas, mas também entender como variantes destes genes — frutos de mutações presentes em linhagens distintas — influenciam na forma das proteínas. Esta abordagem permite entender qual a importância de determinadas mutações para a capacidade do vírus de causar a doença.
O novo coronavírus, denominado SARS-CoV-2, é um vírus (parasita intracelular que depende de um hospedeiro para manter sua viabilidade e se replicar) de RNA surgido em populações de espécies asiáticas de morcegos, tendo posteriormente desenvolvido a capacidade de infectar e ser espalhado em populações humanas. A doença causada pelo SARS-CoV-2 tem o nome de COVID-19 (do inglês, doença do Coronavírus 19 — em referência ao ano de início da atual pandemia), e pode afetar pessoas de todas as idades, tendendo a causar quadros mais graves em adultos de idade mais avançada e pacientes com comorbidades como hipertensão, diabetes, problemas dos sistemas cardíaco, imunológico e respiratório. Segundo o site do Centro para Controle de Doenças dos Estados Unidos, os principais sintomas do novo coronavírus são:
- Febre e/ou calafrios;
- Tosse;
- Dificuldade respiratória e/ou falta de ar;
- Fadiga;
- Dores musculares ou no corpo;
- Dor de cabeça;
- Perda recente dos sentidos de olfato e/ou paladar;
- Dor de garganta;
- Nariz congestionado e/ou com coriza;
- Náusea ou vômito;
- Diarreia;
O site indica ainda que podem ocorrer outros sintomas, além de indicar a busca de serviços de saúde de emergência ao se constatarem os seguintes sintomas:
- Dificuldade respiratória mais grave;
- Dor persistente e/ou pressão no peito;
- Confusão mental;
- Incapacidade de acordar ou se manter em vigília;
- Pele, lábios ou lâmina ungueal (pele sobre abaixo das unhas) em tom pálido, acinzentado ou azulado, a depender da cor de pele;
Os sintomas acima podem estar relacionados ao desenvolvimento de um quadro respiratório conhecido como Síndrome Respiratória Aguda Grave, que pode ocorrer em doenças pulmonares e está altamente associado ao SARS-CoV-2. O novo coronavírus também pode promover quadros graves com hiperinflamação dos tecidos, inclusive do pulmão, além da invasão de tecidos externos ao sistema respiratório (como o Sistema Nervoso Central e os vasos sanguíneos), resultando em outros quadros de saúde, como a encefalite.
É Importante ressaltar que a COVID-19 pode ocorrer sem manifestação de sintomas, ou apenas com sintomas leves, que são frequentemente confundidos com gripes e resfriados, de forma que uma pessoa aparentemente saudável ou com pouco mal-estar pode contaminar as pessoas à sua volta. Portanto, a adoção de práticas como as descritas abaixo em “Como evitar a infecção e o espalhamento do novo coronavírus?” se faz necessária mesmo na ausência de sintomas mais severos.
As melhores maneiras de impedir a disseminação do novo coronavírus são:
- A adoção do distanciamento social com medidas de restrição de circulação de pessoas, como horários especiais para atividade de setores específicos da economia e a redução ou restrição total de atividades não-essenciais — o chamado lockdown, que consiste na completa paralisação de atividades não-essenciais.
- Uso de máscaras, preferencialmente dos tipos PFF2 ou N95, que têm filtração equivalente. Não havendo possibilidade de aquisição de máscaras deste tipo (ou similares da mesma classe de proteção), máscaras de tecido com duas ou três camadas podem ser usadas, embora não ofereçam proteção similar à das máscaras PFF2. Máscaras devem ser utilizadas corretamente, cobrindo a totalidade do nariz e da boca, para que possam cumprir sua função em reduzir a quantidade de partículas virais que escapam em gotículas de saliva. As máscaras do tipo PFF2/N95 podem ser reutilizadas, com a precaução de que sejam deixadas intocadas em local arejado por pelo menos 3-7 dias entre um uso e outro. Já as máscaras de tecido devem ser bem lavadas com água e sabão e bem enxaguadas e secas entre um uso e outro.
- A lavagem das mãos, a cobertura das vias aéreas com cotovelos ou ombros ao tossir e espirrar, e a precaução de não tocar os olhos, o nariz ou a boca com as mãos sujas também diminuem a chance de contrair a doença. Quando não for possível lavar as mãos com água e sabão, usar álcool líquido ou gel.
Apesar de todas as pessoas estarem em risco de contrair a doença, existem parcelas da população que, devido a condições prévias de saúde, estão em maior risco de desenvolvimento de quadros graves, com necessidade de hospitalização, ventilação mecânica e risco aumentado de morte. O site do Centro para Controle de Doenças dos Estados Unidos lista os seguintes grupos / condições de saúde:
- Pessoas com idade avançada;
- Pessoas vivendo em condição de pobreza, devido à maior ocorrência de comorbidades em idades mais jovens;
- Câncer;
- Sobrepeso e obesidade (especialmente obesidade mórbida);
- Doença renal crônica;
- Quadros pulmonares crônicos como doença pulmonar obstrutiva crônica, asma, doença pulmonar intersticial, fibrose cística e hipertensão pulmonar;
- Demência e condições neurológicas relacionadas;
- Diabetes melitus (tipos 1 e 2);
- Síndrome de Down;
- Condições cardíacas;
- Infecção pelo vírus HIV;
- Estado imunocomprometido;
- Doença hepática;
- Gravidez;
- Anemia falciforme ou talassemia;
- Tabagismo (atual e pregresso);
- Pacientes que passaram por transplantes de órgãos ou células-tronco sanguíneas;
- Pacientes que sofreram derrames ou outros tipos de acidente vascular cerebral que reduza a irrigação sanguínea no cérebro;
- Transtornos de abuso de substâncias (alcoolismo, dependência de opióides, cocaína, etc);
Pessoas com estas condições e quem mora junto a elas devem ter cuidados redobrados, reforçar o isolamento, a higiene e o uso de máscaras, de forma a reduzir a probabilidade de infecção ao mínimo risco possível.
A preocupação quanto ao contágio de animais de estimação e da fauna silvestre faz sentido, especialmente se considerado o fato de que o SARS-CoV-2 originou-se em espécies de morcegos asiáticos e “saltou” para a espécie humana (em um processo também conhecido como spillover ou “transbordamento”, que provavelmente envolveu um hospedeiro intermediário). Porém, levando-se em conta que o processo de adaptação para uma nova espécie é relativamente raro, é sempre importante balizar preocupações como esta com base em evidências científicas e estudos que tentem determinar qual a probabilidade real de o SARS-CoV-2 contaminar animais de estimação.
Até o momento, a convivência entre animais como cães e gatos e os seres humanos em meio à pandemia parece não oferecer grandes riscos aos envolvidos. Apesar de haver casos nos quais cães e gatos domésticos testaram positivos para o SARS-CoV-2, a transmissão do vírus para estas espécies parece não comprometer significativamente a saúde dos animais, além de ocorrerem em frequência aparentemente inferior à transmissão de pessoa para pessoa: um artigo produzido com participação de pesquisadores da Rede Genômica Fiocruz verificou a contaminação pelo vírus em proporção equivalente a 31 de cada 100 cães, e de 40 em cada 100 gatos vivendo em residências com pelo menos um paciente humano com o novo coronavírus. Em comparação, um estudo do Centro para Controle de Doenças dos Estados Unidos (CDC) concluiu que a doença acomete 53 pessoas de cada 100 que vivem junto com um paciente com a doença. Uma consideração adicional em relação às taxas de contágio em cães e gatos é que o estudo citado foi realizado com um número relativamente pequeno de animais, o que limita um pouco as conclusões possíveis a respeito do quão comum é a infecção destes animais pelo novo coronavírus.
Independentemente da precisão das taxas observadas em relação ao risco real, uma das recomendações do artigo que se mantém forte é a de que famílias que possuam cães e especialmente gatos, que parecem estar em maior risco, tomem a precaução de evitar contato próximo entre membros da família com casos confirmados ou suspeitos de COVID-19 e os animais, assim como fariam em relação a outros membros humanos da família. É de particular importância que não se permita que os animais durmam na mesma cama que os pacientes acometidos pela doença. Outra consideração importante é a de que animais castrados parecem estar em um risco ligeiramente mais alto do que os não-castrados.
A infecção no caminho contrário, ou seja, de animais de estimação para seres humanos saudáveis, parece não ocorrer. Desta forma, não há motivos, pelo menos por hora, para se temer o contágio a partir de gatos e cães que eventualmente exibirem sintomas ou testarem positivo para o SARS-CoV-2.
Além de gatos, outra espécie domesticada mais expressivamente acometida pelo novo coronavírus são os visons, criados em fazendas em diversos países para abate e confecção de produtos como casacos e luvas de pele. Porém, como no Brasil a criação de animais para extração de pele não é permitida, este não é um problema de saúde coletiva por aqui.
A princípio, pessoas recém-recuperadas de sintomas da COVID-19 ainda podem transmitir a doença por alguns dias. Embora o período de tempo varie de indivíduo para indivíduo — e possivelmente entre variantes do vírus — as recomendações são para que um período de quarentena seja observado antes que pacientes convalescentes possam entrar em contato com outras pessoas. Segundo o site do Centro para Controle de Doenças, pacientes com sintomas leves devem esperar um mínimo de 10 dias contados a partir do início dos sintomas para encontrar outras pessoas. O período de 10 dias também é recomendado para pacientes assintomáticos que tenham testado positivo para o vírus. Nestes casos, os 10 dias são contados a partir da data do teste. Já para pacientes graves ou imunocomprometidos, o período de quarentena deve ser maior, podendo chegar a 20 dias.
Segundo uma norma produzida pelo Hospital Universitário Walter Cândido, da Universidade Federal do Ceará, recomendações adicionais são as de que pacientes que apresentaram sintomas — independente do estado de imunossupressão — mantenham o isolamento para além destes 10 ou 20 dias, até que apresentem 24h sem febre e outros sintomas, na ausência de uso de antitérmicos. Adicionalmente, a norma recomenda que pacientes recém-transplantados (tanto medula óssea quanto transplantes sólidos) ou se preparando para receber um transplante com o uso de medicação imunossupressora tenham um teste não-detectável de RT-PCR após 20 dias desde o início dos sintomas para que possam sair de isolamento.
É importante levar em conta que existem relatos de eventos isolados em que partículas virais ativas, inclusive dotadas de capacidade de replicação, foram isoladas em pacientes pelo menos 18 dias após o aparecimento dos sintomas.
Variantes de um vírus são resultado de mutações que ocorrem naturalmente durante o processo de replicação — tanto em vírus quanto em organismos vivos como o ser humano, outros animais, plantas e microrganismos. Em uma situação como a da presente pandemia, em que um mesmo vírus se espalha por diversas regiões geográficas, é esperado que novas mutações apareçam independentemente em cada localidade. Quando a circulação de pessoas não é controlada, essas variantes podem ainda ser transportadas e se estabelecer em outras regiões.
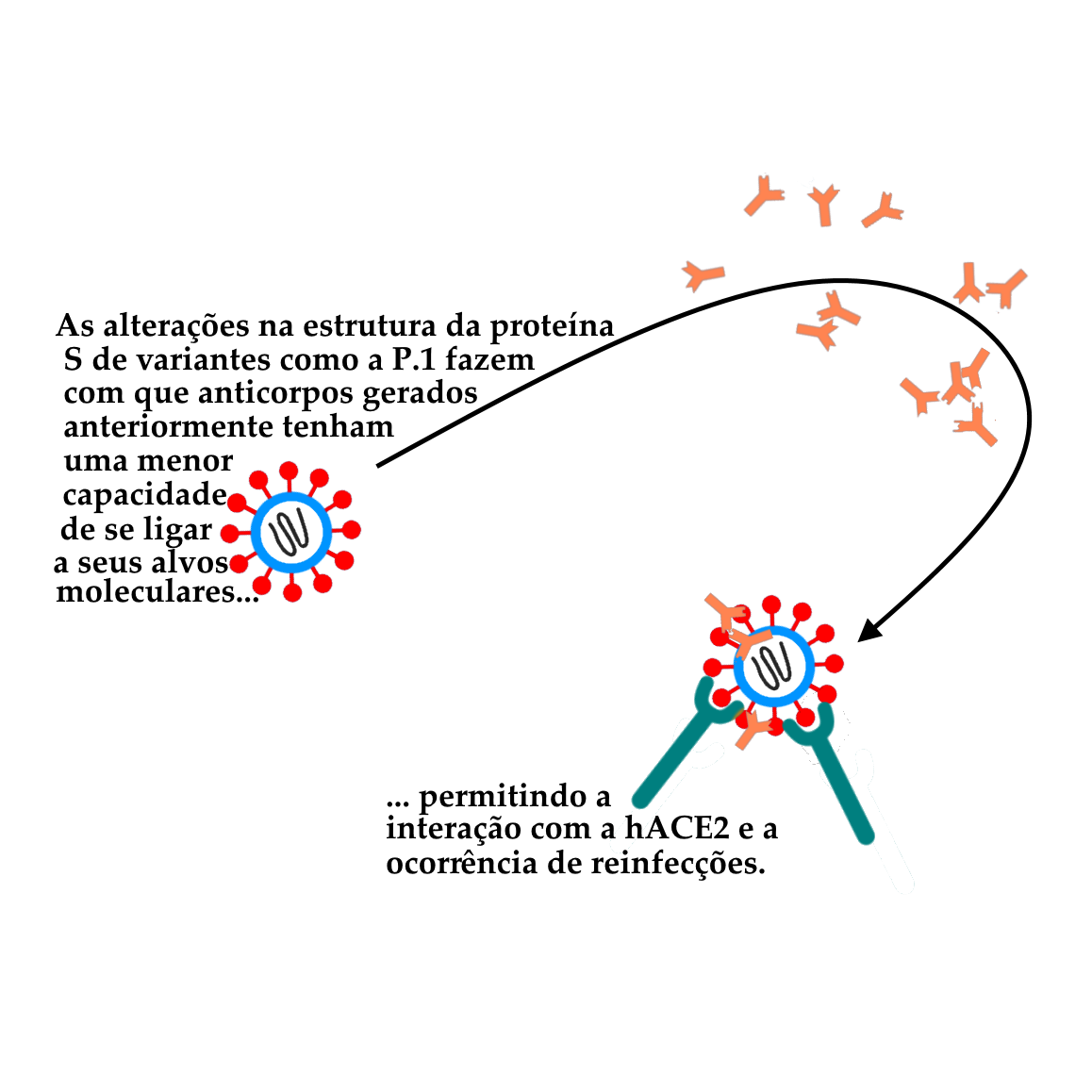
Variantes, tanto em seu local de origem quanto em novas localidades alcançadas, tendem a continuar passando por eventos de mutação e se ramificar em novas entidades. Quando uma mutação confere uma vantagem evolutiva às variantes que a carregam, ela tende a se estabilizar ou se expandir na população, fazendo com que estas variantes se tornem aos poucos mais comuns do que aquelas sem esta mutação. Esse parece ser o caso das mutações na proteína S (envolvida na invasão de células pelo novo coronavírus) acumuladas por variantes como a P.1, que permitem o escape da resposta imunológica desenvolvida após uma primeira infecção, permitindo casos de reinfecção pelo vírus. A melhor maneira de impedir o surgimento de novas variantes é controlar o espalhamento da doença, já que cada novo evento de infecção é uma janela para o surgimento de mutações. Desta forma, uma campanha de imunização idealmente deve ser conduzida de forma a atingir uma cobertura vacinal satisfatória (pelo menos 70% da população) o mais rápido possível, uma vez que uma população parcialmente imunizada pode favorecer a evolução do vírus na direção de variantes capazes de escapar aos mecanismos de defesa da parcela da população imune à doença.
Até o momento, foi constatado nas pesquisas da Rede Genômica e de outros grupos de pesquisa ao redor do mundo que mutações nos genes da glicoproteína Spike (proteína S) do novo coronavírus SARS CoV-2 estão associadas a variantes com grande capacidade e se tornarem dominantes, como a P.1 (variante de Manaus), a B.1.1.7 (variante do Reino Unido) e a B.1.351 (variante Sul-Africana). A existência de alterações em diferentes regiões da proteína S nestas amostras parece estar relacionada a uma menor capacidade de neutralização por parte de anticorpos de pacientes que já tiveram a doença (uma vez que estes anticorpos teriam sido gerados para reconhecer e neutralizar versões da proteína S de outras cepas, que não continham essas alterações). Algumas mutações específicas vêm aparecendo de forma independente em várias VOCs e VOIs, tais como as nas posições N501, E484, K417, N452 e T478 da proteína Spike.
Acredita-se que a capacidade de circular tanto entre pessoas que nunca tiveram contato com o SARS-CoV-2 quanto entre aquelas que já tiveram a doença (reinfecções), aliado a uma carga viral mais alta, é um importante fator para a dominância observada para a variante P.1 em várias regiões do Brasil, como constatado por pesquisadores da Rede Genômica Fiocruz.
As medidas de proteção a serem observadas em relação às novas variantes são as mesmas que protegem de outras linhagens do novo coronavírus, descritas na resposta à pergunta “Como evitar a infecção e o espalhamento do novo coronavírus?” acima.
Por ser uma doença nova, ainda há muitos aspectos a serem compreendidos a respeito da COVID-19, e a relação de seu causador com o sistema imunológico humano ainda traz muitas perguntas à comunidade científica. A princípio, os mecanismos da imunidade adquiridos após uma infecção pelo SARS-CoV-2 mantêm sua função de impedir novas infecções por pelo menos alguns meses, sendo importante o desenvolvimento de vacinas para garantir uma maior chance de imunidade de longo prazo.
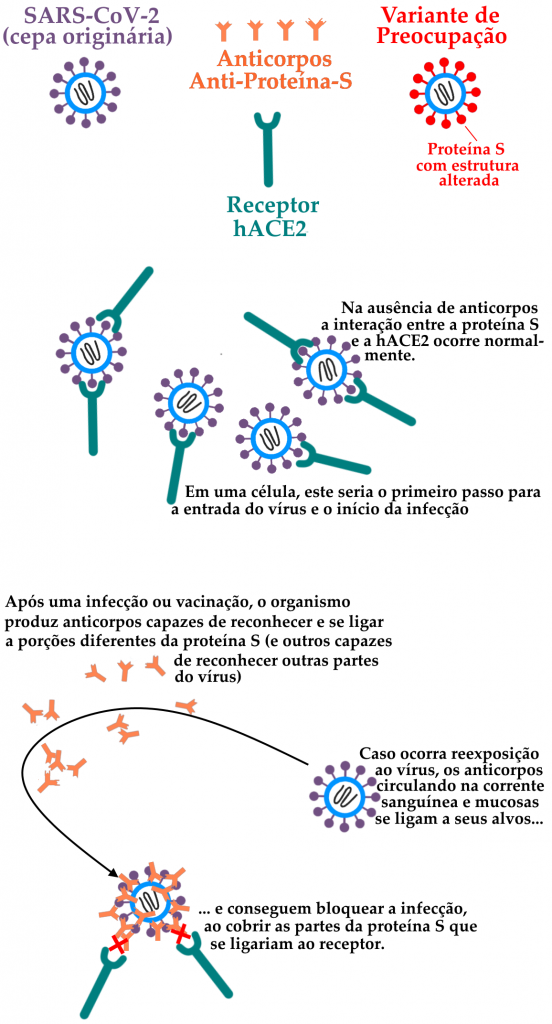
O surgimento de variantes como a P.1, que carregam alterações relevantes na glicoproteína Spike (proteína S, através da qual o vírus consegue se ligar às células hospedeiras e invadi-las, causando infecção) foi responsável por diversos casos comprovados de reinfecção, pois estas alterações diminuem a ação dos anticorpos sobre esta proteína. Estima-se ainda que esse o número real de reinfecções seja bem maior, devido à dificuldade técnica de comprovar esses casos. A menor eficiência dos dos anticorpos frente a essas mutações não consegue evitar completamente que a proteína Spike das variantes possa se ligar ao receptor presente nas células humanas, ocasionando uma nova infecção.
Até o momento (março-2022), as evidências apontam para uma capacidade das vacinas atualmente utilizadas de gerar anticorpos capazes de neutralizar parcialmente o novo coronavírus, incluindo as variantes de preocupação em circulação no momento, prevenindo casos graves da doença. As variantes de preocupação, porém, demonstram-se capazes de infectar mesmo pessoas vacinadas, embora a imunização tenda a proteger de internações e óbitos.
Apesar de, até o momento presente, as vacinas serem aparentemente capazes de proteger contra a infecção grave por novas variantes (ver resposta à pergunta “Quem já recebeu a vacina contra o coronavírus está imune às variantes?” acima) a circulação desenfreada do novo coronavírus em populações parcialmente expostas ao vírus ou parcialmente vacinadas favorece o surgimento de cada vez mais variantes do SARS-CoV-2. A possibilidade de que surjam variantes capazes de escapar completamente à resposta imunológica desencadeada por vacinas é um risco a ser encarado com muita seriedade, uma vez que este evento poderia significar um prolongamento da pandemia.
A Fiocruz trabalha para o combate à pandemia de SARS-CoV-2 principalmente através da pesquisa em diversas frentes de investigação. As pesquisas desenvolvidas pela Fundação como um todo vão desde a compreensão do genoma e das características virais até o estudo da resposta imunológica ao vírus, teste de terapias e desenvolvimento de imunizantes, e muitas destas fazem parte de programas internacionais de cooperação em pesquisa. Apesar do foco em pesquisa científica, no Rio de Janeiro a Fiocruz construiu também um Centro Hospitalar para atender a pacientes graves da COVID-19.
O trabalho da Rede Genômica Fiocruz não envolve coleta direta de material, e os grupos de pesquisa não possuem profissionais da enfermagem ou licença para coleta de material. Todas as amostras utilizadas para extração e sequenciamento do genoma são coletadas por profissionais e saúde, em Hospitais, Clínicas e Unidades Básicas de Saúde e encaminhadas aos Laboratórios Centrais (Lacen) de cada Unidade Federativa. É através da parceria com os Lacens que a Rede Genômica Fiocruz (e institutos de pesquisa parceiros) tem acesso ao estudo do genoma destas amostras.
Não. Conforme explicado na resposta à pergunta “A Rede Genômica Fiocruz realiza testes para diagnosticar se estou com o coronavírus e identificar por quais variantes?” acima, a Rede Genômica Fiocruz não realiza coleta ou processamento de amostras ofertadas diretamente por pacientes, devendo as amostras serem encaminhadas pelas unidades de saúde, institutos de pesquisa biomédica e os Lacens de cada Unidade Federativa.
A elaboração e testagem de vacinas não está entre os objetivos de pesquisa e atribuições da Rede Genômica Fiocruz. A Fundação Oswaldo Cruz é um dos mais importantes institutos de pesquisa biológica e biomédica do mundo, de forma a haver grupos de pesquisa e redes de trabalho com experiência nos mais diversos assuntos. No contexto da nova pandemia, estes laboratórios e redes de colaboração atuam em múltiplas frentes de enfrentamento do novo coronavírus, de acordo com a experiência e conhecimento técnico de cada equipe. Estas pesquisas, mesmo que inicialmente realizadas de maneira independente e com focos distintos, podem eventualmente informar umas às outras.
Por exemplo, embora a Rede Genômica não tenha participado dos testes clínicos de vacinas e não atue em sua produção, os estudos a respeito de novas variantes e dos mecanismos que permitem seu escape da resposta imunológica são importantes para acompanhar os efeitos da vacinação. Estes estudos podem ainda informar, no futuro, a necessidade e os alvos para o desenvolvimento de novas vacinas, a serem oferecidas à população nos anos seguintes ou caso apareçam variantes capazes de escapar completamente do efeito protetor dos imunizantes atuais.
A origem exata do Sars CoV-2 ainda não está elucidada e o tema continua sendo investigado. Existem fortes evidências que apontam para uma origem animal. Comparações entre o genoma do vírus obtidos em pacientes e outros coronavírus revelam uma proximidade com o RatG13, encontrado em morcegos, que provavelmente infectou primeiro um hospedeiro intermediário e eventualmente pulou para humanos em ambientes onde há contato próximos entre as espécies, como mercado de animais ou cavernas. Ademais, não existe na estrutura genômica do SARS-CoV-2 nenhum vestígio de manipulação do material genético do vírus. Esta hipótese foi verificada por diferentes grupos de pesquisa ao redor do mundo, que não encontraram evidências que sustentem esta hipótese.
Especialmente frente ao fato de que o genoma do SARS-CoV-2 é muito próximo ao de outros coronavírus, como o SARS-CoV e o MERS-CoV, ambos vírus de ocorrência natural que já causaram surtos de menores proporções, em diversos momentos na história humana recente, a ideia de que o novo coronavírus veio de um laboratório — e não dos mesmos processos naturais que geraram estes “parentes” — parece improvável.