
Here we have gathered some of the most common questions regarding the novel coronavirus pandemic. The focus of this section is on genomic surveillance, but other important topics that may generate doubt in the population are also covered. The content of this page is under constant revision and new questions and answers will be added over time.
FREQUENTLY ASKED QUESTIONS
The Fiocruz Genomics Network is a set of research groups from several Units of the Oswaldo Cruz Foundation, present in all regions of Brazil, working in coordination to study the genetics and genomics of microorganisms relevant to human health.
The Network has been working on sequencing and studying the genome of SARS-CoV-2, the coronavirus that causes COVID-19, as part of the global effort to track the virus spread across different regions of the world, with a focus on identifying and tracking its genetic mutations and the emergence of new lineages. For more information about the Network and its partner institutions, please visit the “The Network” page of our website.
The high throughput genome sequencing performed on samples of the novel coronavirus (SARS-CoV-2), in addition to allowing the circulation monitoring of different lineages of the virus, the detection of patterns in the emergence of new variants and their spread in relation to other lineages, and the identification of variants not yet described, also allows the study of relevant mutations of the virus.
Understanding how the virus causing Covid-19 spreads between different regions, and monitoring the prevalence of its different genetic lineages, is a process of great relevance to strategic planning to combat the pandemic. These data are of great importance for developing specific diagnostic tests for new variants, updating vaccines, and designing public health measures to contain the disease.
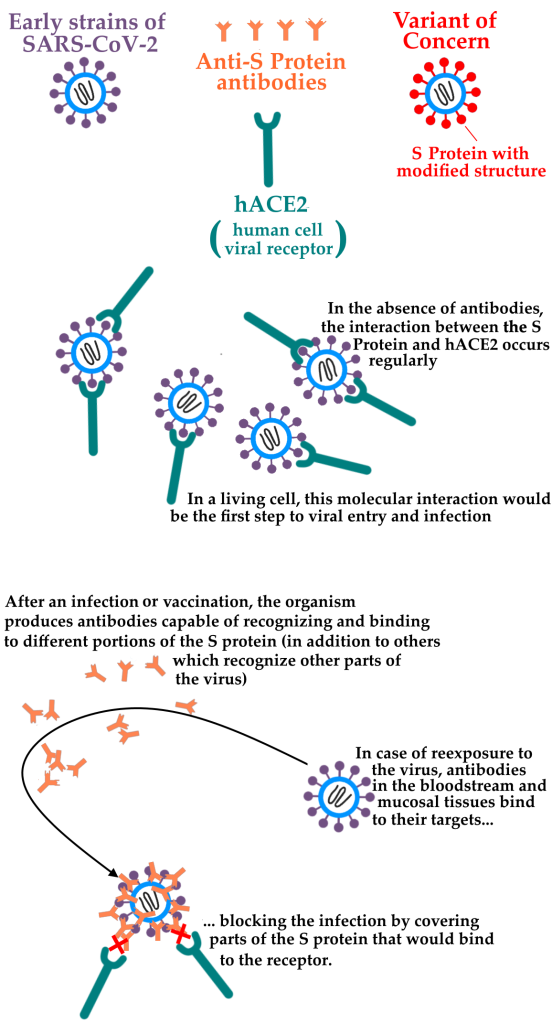
The study of genetic changes such as those in the Spike protein structure – through which the virus enters host cells – is also important for understanding the risks associated with the circulation of these mutant variants. This knowledge allows the identification of mutations that can serve as “warning signals” by appearing independently in distinct locations, such as those at position E484 and N501 of the Spike protein.
The Genomics Network is the result of the coordinated work of laboratories and research groups in 12 units of Fiocruz, which already traditionally carried out studies with the genomics of the most diverse organisms of interest to human health before the Covid-19 pandemic. Thus, the sequencing and analysis of the genetic material of organisms that cause diseases such as Leishmania, respiratory viruses such as influenza (flu) and respiratory syncytial virus, and other diseases such as dengue, measles, and schistosomiasis were already part of the laboratories routine that make up the network before the current public health emergency.
As part of the SARS-CoV-2 study, the Genomics Network studies the genetic sequences of coronavirus samples collected throughout Brazil, feeding the database of the international GISAID initiative and identifying the variant lineages of the virus circulating in the different regions of the country.
The research groups that make up the Genomics Network have been working for years to study the most diverse aspects of the genome of organisms that cause infectious diseases. Thanks to the research of laboratories that are now part of the network, we better understand the pathogenesis mechanisms – that is, the mechanisms through which an organism or virus causes a clinical picture – the spread, evolution, and genome of the agents that cause diseases that are relevant to the Brazilian population.
These are included studies focused on arbovirosis such as dengue and zika, and tropical parasitosis such as Leishmaniasis and Chagas disease. Mapping locations where these diseases occur, jointly with genomic analysis of samples at each site, aids in detecting higher risk areas, formulating control and prevention strategies, and understanding the evolution of causative agents. The genome study of these pathogens is also important for a more detailed understanding of the mechanisms of each disease, which opens doors for the development of therapeutic and/or prophylactic approaches (such as vaccines) to combat them and restore health to affected patients and their families.
Since 2020, with the formalization of the Genomics Network, this research is focused on the novel SARS-CoV-2 coronavirus, and on understanding multiple aspects of the current pandemic caused by this new pathogen.
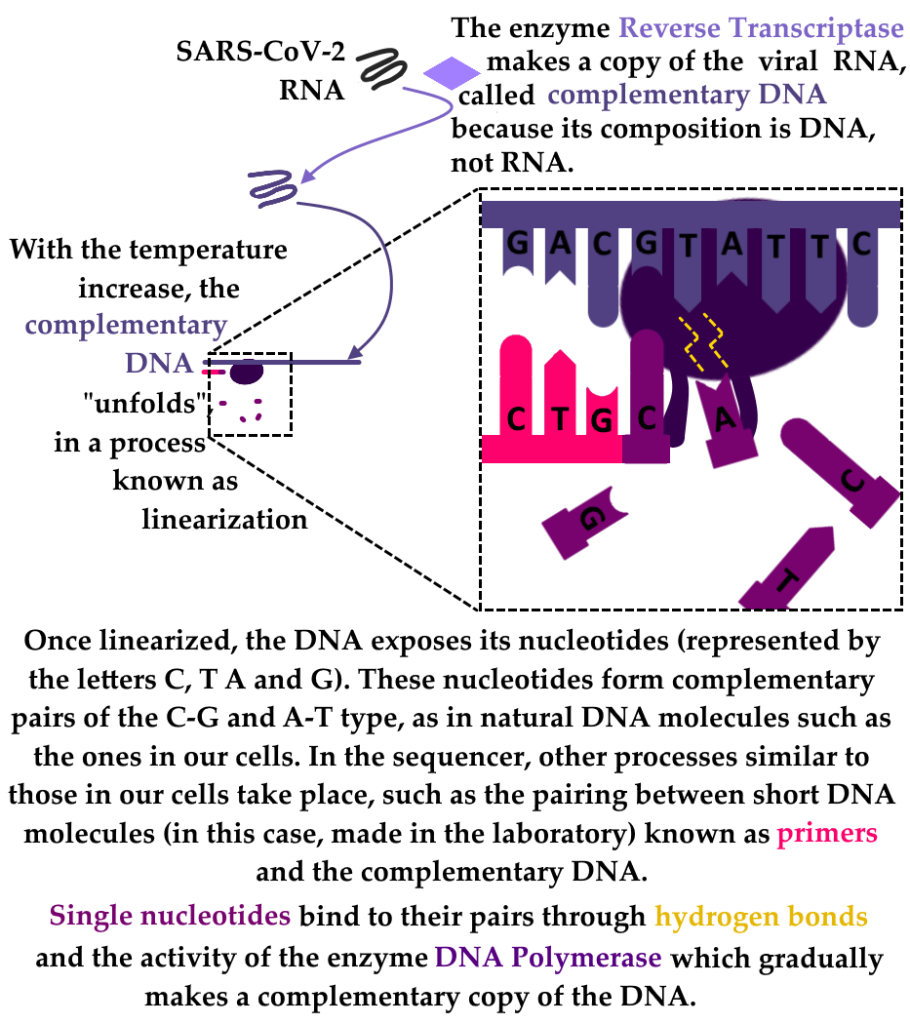
Gene sequencing is a technique based on chemical reactions that already happen naturally in cells when genetic material is replicated. By associating the enzymes used in this duplication with a technology involving fluorescence, sequencing techniques make it possible to learn part or all of the information present in the genetic code of an organism. It therefore allows you to identify genes, predict their products (proteins, interference RNAs, ribozymes, etc.), and compare sequences from different organisms.
With the temperature increase, the complementary DNA “opens”, in a process known as linearization | The Reverse Transcriptase enzyme makes a copy of the viral RNA, called complementary DNA, because its composition is DNA, not RNA. | Once linearized, the DNA exposes its nucleotides (represented by the letters C, T A and G). These nucleotides form complementary pairs of the C-G and A-T type, even in natural DNA molecules such as that of our cells. In the sequencer, other processes similar to those in our cells take place, such as the pairing between short DNA molecules (in this case, made in the laboratory) known as primers and the complementary DNA. Single nucleotides bind to their pairs through hydrogen bridges and the action of the DNA Polymerase enzyme, which gradually builds a complementary copy of the DNA.
From this comparison, between sequences of older SARS-CoV-2 samples and sequences of more recent samples, it is possible to identify the commonalities and points of divergence between their genomes. This analysis allows tracing of evolutionary lines and the analysis of kinship between variants. Thus, virus samples that have the same origin, and differentiated from the same ancestor, are classified in the same lineage, and it can also be understood how one lineage gives rise to others.
To perform the sequencing, a smaller amount of nucleotides with some tagging (usually fluorescent) are used along with the conventional nucleotides. It is also common for these nucleotides to have chemical changes that block the construction of the complementary copy of DNA.
When the DNA Polymerase inserts one of these changed nucleotides, it has no way to add the next one, and the synthesis of this complementary copy stops. As this synthesis process happens thousands to millions of times on a sequencer, it is possible for research groups to unravel the genome sequences being studied by analyzing which tagged nucleotides have been placed in each portion of complementary DNA copies, either by reading fluorescence or by other methods, depending on the technology available.
In any large-scale epidemic event, especially those characterized by a rapid spread of the etiologic agent, case monitoring is important to follow the dynamics of the epidemic. The genetic profile of the virus provides information about its spread from a geographical and temporal standpoint, revealing its main transmission routes. In other words, the genome is the pathogen identity and reveals where and when it spread.
One year after the emergence of Sars CoV-2, genetic sequencing takes on an even more central role in fighting the pandemic due to the constant emergence of new variants of the virus. These are classified as “variants of interest” (those that should be closely monitored, but are less likely to be associated with more severe cases or higher infectivity) and “variants of concern” (those that are likely to have substantial implications for public health). The study of mutations also allows a better understanding of the pathogenicity mechanisms, i.e., the genes associated with the emergence of the disease, its severity, or its ability to infect new hosts.

How does genome annotation work? What answers can the technique give us about the new lineages?
Genome annotation consists of analyzing the data generated after sequencing, that is, the computer-aided detailed study of the genetic information sequences. The annotation process involves the identification of structures that mark regions of the genome, such as the beginning and end of genes, the sequences involved in the regulation of expression (activation or deactivation), and eventually regions that are not expressed. Annotation is therefore the interpretation and organization of genetic information. It can help, for example, in comparing different versions of the same gene, present in samples from different lineages, and understand what are the changes in these genes (and in nearby sequences, which can influence their expression). Thus, by unraveling the sequences direction, annotation is an indispensable step in the study of the novel coronavirus genome, and in the classification of samples into lineages.
Genome reconstructed on computer | With computer aid, it is possible to analyze the sequencing result. In general, this process involves reconstructing the genome by comparing several portions and identifying overlapping sequences. It is a lot like putting a puzzle together. | The use of the computer also allows a process called genome “annotation”. This step is when the relevant information in the genome is looked at closely: the identification of genes and their regulation mechanisms takes place in the annotation, by studying the nucleotide sequences and comparing them with other known sequences, deposited in international databases.
Monitoring the spread of new variants allows the risk of each to be assessed with regard to spread during a pandemic. Combined with monitoring the rate of social isolation and situations that promote the relaxation of care (for example: reopening of some economic sectors, extended holidays, commemorative dates, and political events), variant monitoring allows the identification of key events in its spread, in order to make it possible to understand how a variant reaches a new locality. This work also helps to evaluate the strategies effectiveness to combat the epidemic, and can inform awareness campaigns about the best actions to be taken to contain the disease advance.
Understanding an infectious agent genome – whether a virus, bacterium, fungus, or parasite – enables a number of approaches to fighting the disease it causes. As far as the development of treatments is concerned, the genetic sequences analysis brings information about the pathophysiology of the virus, identifying possible targets for therapeutic intervention.
Additionally, the study of genes involved in pathogenesis can aid in the development of new therapies through another approach: three-dimensional molecular modeling. Using bioinformatics tools, it is possible to build models of important proteins, such as Spike from the novel coronavirus, involved in host cell invasion. Based on the study of the gene sequences responsible for the expression of this protein, it is possible not only to predict its structure and develop molecules that block its binding to receptors present on human cells, but also to understand how variants of these genes – the result of mutations present in different lineages – influence the proteins shape. This approach allows to understand how important certain mutations are to the virus’ ability to cause disease.
The novel coronavirus, called SARS-CoV-2, is an RNA virus (an intracellular parasite that depends on a host for viability and replication) that emerged in populations of Asian bat species and subsequently developed the ability to infect and spread to human populations. The disease caused by SARS-CoV-2 is called COVID-19 (Coronavirus 19 disease – in reference to the year when the current pandemic started). It can affect people of all ages, but tends to be more severe in older adults and patients with comorbidities such as hypertension, diabetes, and heart, immune, and respiratory system problems. According to the U.S. Centers for Disease Control’s website, the main symptoms of the novel coronavirus are:
- Fever and/or chills;
- Cough;
- Difficulty breathing and/or shortness of breath;
- Fatigue;
- Muscular or body aches;
- Headache;
- Recent loss of the senses of smell and/or taste;
- Sore throat;
- Runny Nose and/or Nasal Congestion
- Nausea or vomiting;
- Diarrhea;
The site also indicates that other symptoms may occur, and also indicates seeking emergency health services when the following symptoms are found:
- More severe breathing difficulty;
- Persistent chest pain and/or pressure;
- Mental confusion;
- Inability to wake up or stay awake;
- Pale, grayish, or bluish skin, lips, or nail beds (skin under the fingernails), depending on the skin color;
The above symptoms may be related to the development of a respiratory condition known as Severe Acute Respiratory Syndrome, which can occur in lung diseases and is highly associated with SARS-CoV-2. The novel coronavirus can also promote severe conditions with hyperinflammation of the tissues, including the lung, and invasion of tissues outside the respiratory system (such as the Central Nervous System and blood vessels), resulting in other health conditions such as encephalitis.
Importantly, COVID-19 can occur without symptoms or with only mild symptoms, which are often mistaken for colds and flu, so that an apparently healthy person with little discomfort can infect those around them. Therefore, the adoption of practices such as those described below in “How to prevent infection and the spread of the novel coronavirus?” is necessary even in the absence of more severe symptoms.
The best ways to prevent the spread of the novel coronavirus are:
The adoption of social distancing with measures to restrict the circulation of people, such as special hours for activities in specific sectors of the economy and the reduction or total restriction of non-essential activities – the so-called lockdown.
Use of masks, preferably PFF2 or N95 types, which have equivalent filtration. If it is not possible to buy masks of this type (or similar masks of the same protection class), fabric masks with two or three layers can be used, although they do not offer protection similar to that of PFF2 masks. Masks must be worn correctly, covering the entire nose and mouth, so that they can fulfill their function in reducing the amount of viral particles that escape in saliva droplets. The PFF2/N95 masks can be reused, with the precaution that they are left untouched in a ventilated place for at least 3-7 days between one use and another. Fabric masks, on the other hand, should be well washed with soap and water and thoroughly rinsed and dried between each use.
Hand washing, covering the airways with elbows or shoulders when coughing and sneezing, and taking the precaution of not touching the eyes, nose, or mouth with dirty hands also decrease the chance of contracting the disease. When it is not possible to wash your hands with soap and water, use liquid or gel alcohol.
Although all people are at risk of contracting the disease, there are portions of the population that, due to previous health conditions, are at greater risk of developing severe conditions, with the need for hospitalization, mechanical ventilation, and increased risk of death. The U.S. Centers for Disease Control’s website lists the following groups / health conditions:
- People with advanced age;
- People living in poverty, due to the higher occurrence of comorbidities at younger ages;
- Cancer;
- Overweight and obesity (especially morbid obesity);
- Chronic kidney disease;
- Chronic lung conditions such as chronic obstructive pulmonary disease, asthma, interstitial lung disease, cystic fibrosis, and pulmonary hypertension;
- Dementia and related neurological conditions;
- Diabetes mellitus (types 1 and 2);
- Down’s Syndrome;
- Cardiac conditions;
- HIV infection;
- Immunocompromised state;
- Liver disease;
- Pregnancy;
- Sickle cell anemia or thalassemia;
- Smoking (current and past);
- Patients who have undergone organ or blood stem cell transplants;
- Patients who have suffered strokes or other types of cerebrovascular accident that reduce the blood supply to the brain;
- Substance abuse disorders (alcoholism, opioid addiction, cocaine addiction, etc.);
People with these conditions and those who live near them should take extra care, reinforce isolation, hygiene, and the use of masks, in order to reduce the likelihood of infection to the lowest possible risk.
The concern about infecting pets and wildlife makes sense, especially considering the fact that SARS-CoV-2 originated in Asian bat species and “jumped” into the human species (in a process also known as spillover, probably involving an intermediate host). However, given that the adaptation process to a new species is relatively rare, it is always important to base concerns like this on scientific evidence and studies that attempt to determine the actual likelihood of SARS-CoV-2 infecting pets.
So far, the coexistence between animals such as dogs and cats and humans in the midst of the pandemic does not seem to offer any major risks to those involved. Although there are cases in which domestic dogs and cats have tested positive for SARS-CoV-2, the transmission of the virus to these species does not seem to significantly compromise the animals’ health, besides apparently occurring at a lower frequency than person-to-person transmission: a paper produced with participation of researchers from the Fiocruz Genomics Network verified contamination by the virus in a proportion equivalent to 31 out of every 100 dogs, and 40 out of every 100 cats living in households with at least one human patient with the novel coronavirus. In comparison, a study by the U.S. Centers for Disease Control (CDC) found that the disease affects 53 out of every 100 people who live together with a patient with the disease. An additional consideration regarding the infection rates in dogs and cats is that the cited study was conducted with a relatively small number of animals, which limits somewhat the possible conclusions regarding how common the infection of these animals by the novel coronavirus is.
Regardless of the accuracy of the observed rates relative to actual risk, one of the recommendations in the paper that remains strong is that families with dogs and especially cats, which appear to be most at risk, take the precaution of avoiding close contact between family members with confirmed or suspected COVID-19 cases and their animals, as they would with other human family members. It is of particular importance that animals are not allowed to sleep in the same bed as patients affected by the disease. Another important consideration is that castrated animals seem to be at a slightly higher risk than non-castrated ones.
Infection in the reverse path, i.e., from pets to healthy humans, does not seem to occur. Thus, there is no reason, at least for now, to fear infection from cats and dogs that eventually exhibit symptoms or test positive for SARS-CoV-2.
In addition to cats, another domesticated species most significantly affected by the novel coronavirus are minks, which are raised on farms in several countries for slaughter and to make products such as fur coats and gloves. However, since in Brazil the breeding of animals for their fur extraction is not allowed, this is not a collective health problem here.
The best way to protect yourself, until vaccination coverage reaches a significant percentage of the population, is to follow the guidelines of not promoting crowds (parties, barbecues, family gatherings, etc.), leave home only when necessary, and follow the recommendations for mask use and hand hygiene best explained in the answer to the question “How to avoid infection and the spread of the novel coronavirus?
Variants of a virus are the result of mutations that occur naturally during the replication process – both in viruses and in living organisms such as humans, other animals, plants, and microorganisms. In a situation like the present pandemic, in which the same virus spreads through several geographical regions, it is expected that new mutations will appear independently in each locality. When the movement of people is not controlled, these variants can still be transported and establish themselves in other regions.
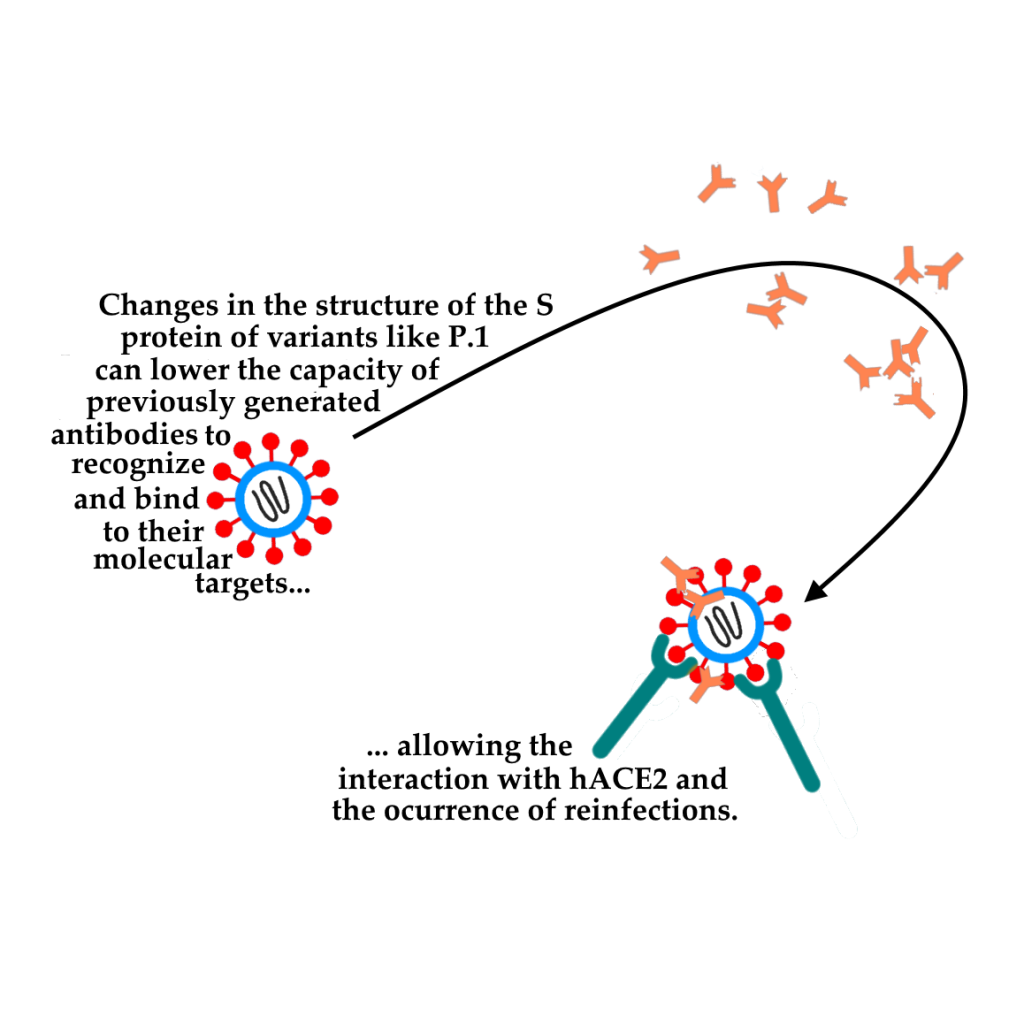
Variants, both in their place of origin and in new locations reached, tend to continue to undergo mutation events and branch out into new entities. When a mutation confers an evolutionary advantage to the variants that carry it, it tends to stabilize or expand in the population, causing these variants to gradually become more common than those without this mutation. This seems to be the case with mutations in the S protein (involved in the cells invasion by the novel coronavirus) accumulated by variants such as P.1, which allow the immune response developed after a first infection to escape, allowing reinfection cases by the virus. The best way to prevent the emergence of new variants is to control the disease spread, since each new infection event is a window for the emergence of mutations. Thus, an immunization campaign should ideally be conducted in order to reach a satisfactory vaccination coverage (at least 70% of the population) as soon as possible, since a partially immunized population may favor the evolution of the virus towards variants capable of escaping the defense mechanisms of the immune population.
Variant detection and monitoring is still very new, so there is a lot to learn about their differences in clinical terms. So far, no new symptoms associated with the known variants have been found. It is known, however, that some of these variants – such as the strain from Manaus, Gama (P.1), the strain in South Africa, Beta (B.1.351), in the UK, Alpha (B.1.1.7), and the strain in India, Delta (B.1.617.2), are more transmissible. Moreover, the Gamma strain is associated with a higher viral load in patients, and carries mutations that allow it to escape neutralization by antibodies (causing reinfection) and to have a higher affinity binding to the human cell receptor, hACE2.
As long as the virus remains in circulation, the accumulation of mutations is inevitable. The so-called variant strains are groups of viruses that share one or more mutations that differentiate them from the “original” strains, which are more like those at the beginning of the pandemic.
In order to organize the data generated around the world, the World Health Organization has published a list of variants of the novel SARS-CoV-2 coronavirus with relevant mutations. This list is constantly updated and divides the variants that require more cautious monitoring into three categories:
- Variants of Interest (VOI): this is a very sensitive classification, so that variants with the potential to cause health situation worsening do not go unnoticed. It only takes a few mutations in relevant genes for a sample to fit this profile. To be classified as a VOI, a variant must have a non-silent mutation (generating amino acid changes) possibly associated with changes in transmissibility, virulence (ability to generate severe symptoms), epidemiology, or in the ability to be recognized by the immune system, in addition to having caused multiple cases of the disease/community transmission.
- Variants of Concern (VOC): A VOI can be reclassified as a VOC when, in a comparative analysis with other VOIs, its greater transmissibility (or negative effects on the epidemiology of the virus in the population), virulence (more severe clinical presentation) or a decreased effectiveness of public health measures (including social distancing and the currently available diagnostic, vaccine and treatment possibilities) against the variant in question.
- Variants under Monitoring (VUM) is a classification given to mutant SARS-CoV-2 gene profiles that do not necessarily pose risk, but have characteristics that warrant more cautious monitoring. The World Health Organization classifies lineages as VUMs when some of their genetic changes are suspected to have effects on viral characteristics with potential future risk , but without conclusive evidence about the increased risk associated with these mutations based on current knowledge.
So far, research by the Genomics Network and other research groups around the world has found that mutations in the Spike glycoprotein (S protein) genes of the novel SARS-CoV-2 coronavirus are associated with variants with a high capacity to become dominant, such as P.1 (Manaus variant), B.1.1.7 (UK variant), and B.1.351 (South African variant). The existence of changes in different regions of protein S in these samples seems to be related to a lower capacity of neutralization by antibodies from patients who already had the disease (since these antibodies would have been generated to recognize and neutralize versions of protein S from other strains, which did not contain these changes). Some specific mutations have been appearing independently in various VOCs and VOIs, such as those at positions N501, E484, K417, N452, and T478 of the Spike protein.
The ability to circulate both among people who have never had contact with SARS-CoV-2 and those who have already had the disease (reinfections), coupled with a higher viral load, is believed to be an important factor for the dominance observed for the P.1 variant in several regions of Brazil, as found by researchers at the Fiocruz Genomics Network.
The protective measures to be observed in relation to the new variants are the same as those protecting from other lineages of the novel coronavirus, described in the answers to the questions “How do I prevent infection and the spread of the novel coronavirus?” and “In the absence of treatment and widespread vaccination, how can I best protect myself and my family?” above.
Because it is a new disease, there are still many aspects to be understood about COVID-19, and the relationship of its causer to the human immune system still brings many questions to the scientific community. In principle, the immunity mechanisms acquired after infection with SARS-CoV-2 retain their function of preventing new infections for at least a few months, and vaccine development is important to ensure a greater chance of long-term immunity.
The emergence of variants such as P.1, that carry relevant changes in the Spike glycoprotein (S protein, through which the virus is able to bind to host cells and invade them, causing infection) was responsible for several proven cases of reinfection, because these changes decrease the antibodies action on this protein. It is also estimated that the actual number of reinfections is much higher, due to the technical difficulty in proving these cases. The lower efficiency of the antibodies against these mutations cannot completely prevent the variants Spike protein from binding to the receptor present on the human cells, causing a new infection.
To date, although vaccines are apparently able to protect against severe infection by new variants (see answer to the question “Are those who have received the coronavirus vaccine immune to the variants?” above) the rampant circulation of the novel coronavirus in partially exposed or partially vaccinated populations favors the emergence of more and more SARS-CoV-2 variants. The possibility that variants will emerge that are able to completely evade the immune response triggered by vaccines is a risk to be taken very seriously, since this event could mean an extension of the pandemic.
Fiocruz is working to combat the SARS-CoV-2 pandemic primarily through research on several fronts. The research undertaken by the Foundation, as a whole, ranges from understanding the genome and viral characteristics to studying the immune response to the virus, testing therapies, and developing immunizers, and many of these are part of international research cooperation programs. Despite the focus on scientific research, Fiocruz has also built a Hospital Center in Rio de Janeiro to attend to severe COVID-19 patients.
The Fiocruz Genomics Network work does not involve direct material collection, and the research groups do not have nursing staff or a license to collect material. All samples used for genome extraction and sequencing are collected by health professionals in hospitals, clinics, and primary health care units and forwarded to the Central Laboratories (Lacen) in each Federative Unit. It is through the partnership with the Lacens that the Fiocruz Genomics Network (and partner research institutes) has access to study the genome of these samples.
No. As explained in the answer to the question “Does the Fiocruz Genomic Network perform tests to diagnose if I have the coronavirus and identify by which variants?” above, the Fiocruz Genomic Network does not perform collection or processing of samples offered directly by patients, and the samples must be forwarded by the health units, biomedical research institutes, and the Lacens of each Federative Unit.
The development and testing of vaccines is not among the research objectives and tasks of the Fiocruz Genomic Network. The Oswaldo Cruz Foundation is one of the most important biological and biomedical research institutes in the world, so that there are research groups and networks with expertise in the most diverse subjects. In the context of the new pandemic, these laboratories and collaboration networks act on multiple fronts to confront the novel coronavirus, according to the experience and technical knowledge of each team. These surveys, even if initially conducted independently and with distinct focuses, can eventually inform each other.
For example, although the Genomics Network has not participated in the vaccines clinical testing and is not involved in their production, new variants studies and the mechanisms that allow them to escape the immune response are important to monitor the vaccination effects. In the future, these studies may also inform the need and targets for the development of new vaccines, to be offered to the population in the following years or if variants appear that are able to completely evade the protective effect of current immunizers.
The exact origin of SARS-CoV-2 has not yet been clarified and the issue is still being investigated. There is strong evidence pointing to an animal origin. Comparisons between the genome of the virus obtained in patients and other coronaviruses reveal a proximity to RaTG13, found in bats, which probably first infected an intermediate host and eventually jumped to humans in environments where there is close contact between the species, such as animal markets or caves. Furthermore, there is no trace of genetic material manipulation of the virus in the genomic structure of SARS-CoV-2. This hypothesis has been verified by different research groups around the world, who have found no evidence to support this hypothesis.
Especially facing the fact that the genome of SARS-CoV-2 is very close to that of other coronaviruses, such as SARS-CoV and MERS-CoV, both naturally occurring viruses that have caused minor outbreaks at various times in recent human history, the idea that the novel coronavirus came from a laboratory – and not from the same natural processes that generated these “relatives” – seems unlikely.