
The terms in this Glossary, available in alphabetical order, were elaborated based on entries found on the site and in publications of the Fiocruz Genomics Network. The content of this page is under constant revision and new terms will be added over time.
Amplicon
Name given to DNA pieces amplified in PCR reactions. Both reaction products and the sequences used as templates are called “amplicon”.
Bayesian Analysis
Bayesian statistics is an approach to data treatment where, unlike classical statistics (frequentist), prior knowledge about the phenomenon studied is explicitly taken into account in the analyses. This approach can provide more informed analyses by taking into account models built on previous data (e.g., from previous research) rather than the hypothetical distributions used in frequentist statistical tests. From phylogenetic analysis standpoint, Bayesian analysis involves incorporating prior knowledge about groups of organisms (or viruses) to derive conclusions about the relatedness of new samples analyzed – both to each other and to previously known organisms.
Common ancestry
In evolutionary biology, a “common ancestry” is one from which other organisms are derived. This ancestry can take place on different scales, so that it is possible to speak of:
- Whole groups of organisms that are derived from common ancestries (as in the case of birds, which are descended from dinosaurs);
- Species in the same group that share common ancestry. For example, three of the species in the genus Panthera, the lions, leopards, and jaguars, are descended from the same ancestor, which is different from the species that gave rise to tigers and snow leopards. In turn, these two ancestral species also derived from the same common ancestor;
- Lineages within the same species, as in the case of the SARS-CoV-2 lineages that causes COVID-19. From a common viral genome, new lineages were derived by the accumulation of genetic mutations, so that it was possible to group variants into clusters according to their common genetic histories;
Bioinformatics
Bioinformatics is the name given to the many computer applications developed for the study of biology. The uses of bioinformatics range from computer programs for the analysis of genomic sequences and comparison with similar sequences already identified, to three-dimensional modeling of the biomolecules structure such as proteins and sugars. In the Genomics Network context, bioinformatics is used primarily to analyze and compare the genomes of different samples of the novel SARS-CoV-2 coronavirus in order to identify mutations characteristic of the new variants and even detect novel mutations. The use of three-dimensional protein modeling tools is also gaining more space in the Genomics Network, which will allow us to study in depth the Spike protein of the novel coronavirus – which allows its entry into cells – and the mutations effects on the structure and functionality of this protein, with possible increase or decrease of infectivity.
Coronavirus
Group of viruses (subfamily Orthocoronaviridae) discovered in 1937, associated mainly with respiratory diseases in animals, including humans. The current pandemic is caused by the virus called SARS-CoV-2, because of its similarity to the agent that caused the SARS (Severe Acute Respiratory Syndrome) epidemic in 2003.
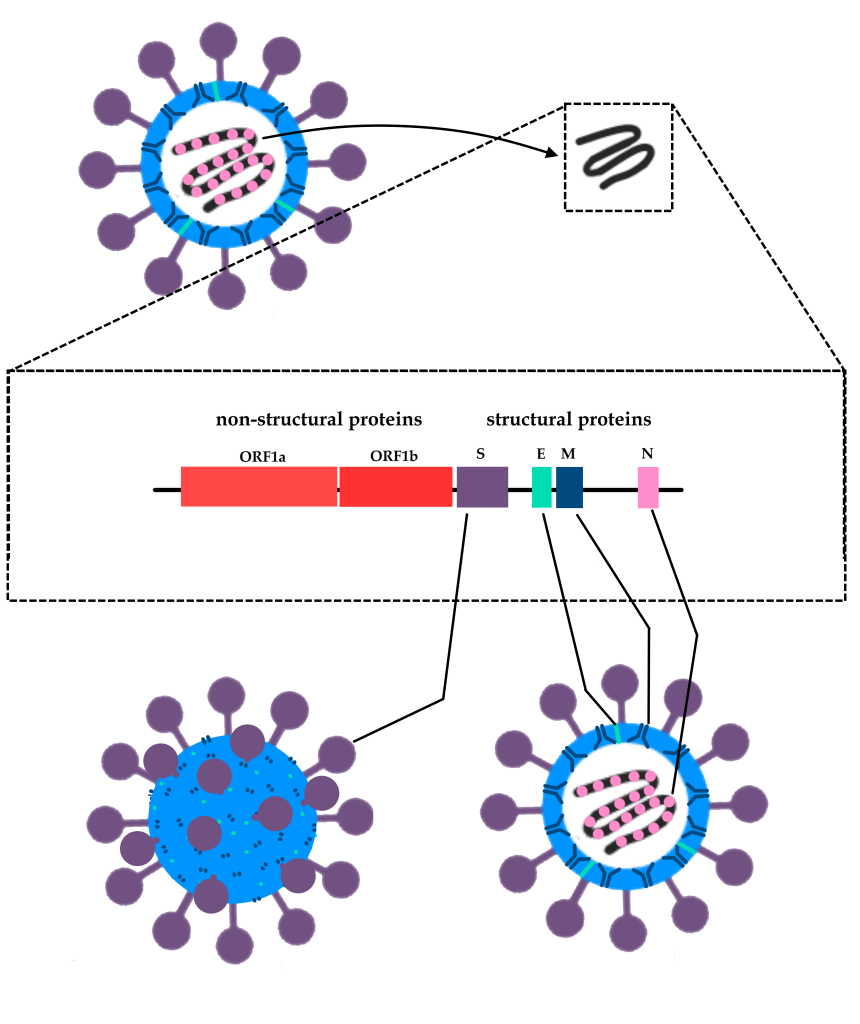
Simplified scheme of the genomic structure of SARS-CoV-2, spotlighting the genes encoding the Spike glycoprotein (S), the envelope protein (E), the membrane protein (M), and the protein that surrounds the RNA within the complete viral particle, forming the nucleocapsid (N). Also prominent are the regions of the genome called ORF1a and ORF1b, where part of the genes for non-structural proteins in the virus are located – that is, they are not part of the viral particle structure. These proteins have diverse functions in infection, such as making copies of the viral RNA inside the cell and redirecting cellular metabolism to turn the cell into a “virus factory.”
Prior to SARS-CoV-2, six other coronaviruses capable of causing infections in humans had been identified, four of these being involved in mild conditions similar to common colds. In addition to these four milder disease profile coronaviruses and SARS-CoV (or SARS-CoV-1), causing the 2003 epidemic characterized by a more severe respiratory picture, the other disease-causing coronavirus in humans is the causative agent of MERS (Middle East Respiratory Syndrome), an epidemic associated with camel breeding and higher mortality rates (37%) than SARS and COVID-19.
The coronavirus enters the cells from the binding of the protein called Spike (or S), with a domain that binds to the angiotensin-converting enzyme 2 – protein that is on the human cells surface and acts as a virus “receptor” – and another domain that causes the fusion between the cell membrane and the viral particle, allowing the entry of the virus genetic material (RNA) and the start of the infection.
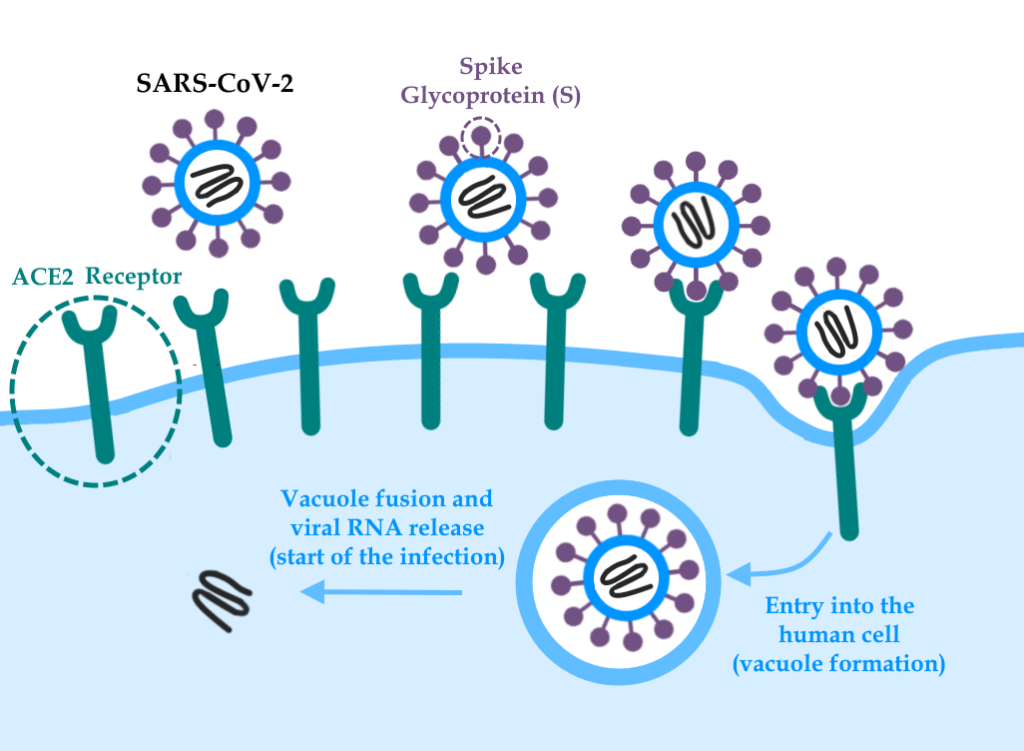 Schematic illustrating the binding between the Spike glycoprotein (S protein) of SARS-CoV-2 and the Angiotensin-Converting Enzyme 2 (ACE-2) present in human cells. After binding, the cell internalizes the viral particle in a vacuole, which then fuses to the viral envelope, releasing the genetic material that will start the infection.
Schematic illustrating the binding between the Spike glycoprotein (S protein) of SARS-CoV-2 and the Angiotensin-Converting Enzyme 2 (ACE-2) present in human cells. After binding, the cell internalizes the viral particle in a vacuole, which then fuses to the viral envelope, releasing the genetic material that will start the infection.
The hyperinflammation characteristic of COVID-19 is one of the main pathogenic mechanisms of the disease, resulting in fever and loss of respiratory function, which in turn can lead to the need for hospitalization and mechanical ventilation in more severe cases. The excess production of cytokines – communicating molecules between cells, involved in the immune mechanisms regulation – associated with inflammatory conditions is directly related to fever and tissue involvement followed by fibrosis, in a phenomenon called “cytokine storm”. The cytokine storm is associated with other viral conditions that have the effects of hyperinflammation as an important part of the pathogenesis, such as severe cases of dengue, for example.
SARS-CoV-2 is believed to have originated in bat species and due to mutations was able to infect and be transmitted among other intermediate mammalian species, eventually including the human species. Analyses of samples circulating among other animal populations place the Malaysian pangolin as a possible intermediary in this process of adaptation to other hosts (also called spillover). The COVID-19 pandemic, with more than 2 million confirmed deaths worldwide as of January 2021, is the largest humanity has experienced since the first H1N1 pandemic of 1917-1918, which caused an estimated 50 million deaths globally.
COVID-19
Name given to the disease caused by the novel coronavirus (called SARS-CoV-2). From English, COrona VIrus Disease 19 (because of the pandemic starting year, 2019). COVID-19 is characterized by high transmissibility and a percentage of asymptomatic or mildly symptomatic cases. After an incubation period of 2 to 14 days, unspecific initial symptoms (cough, fever, olfactory loss, sore throat, headache, fatigue, and eventually diarrhea) may appear), which may be followed by respiratory distress, hypoxia, and severe inflammation of the respiratory tract, including the lungs.
Because it is still a very recent disease, knowledge about COVID-19 still needs further study, especially regarding the long-term effects, as well as comorbidities resulting from SARS-CoV-2 infection, in both symptomatic and asymptomatic people. In rare cases, encephalitis (inflammation of the brain) and the involvement of other tissues can occur, with conditions such as epididymitis and vascular complications appearing in the scientific literature.
Viral genome Evolution
Genome evolution means the sum of the processes of: genetic differences emergence; mutations accumulation; genetic recombination that can occur when the same patient is infected by two different genetic variants; and, finally, the process of natural selection that acts on genetic mutations, favoring those that have some kind of advantage over others, for example, a greater infective capacity.
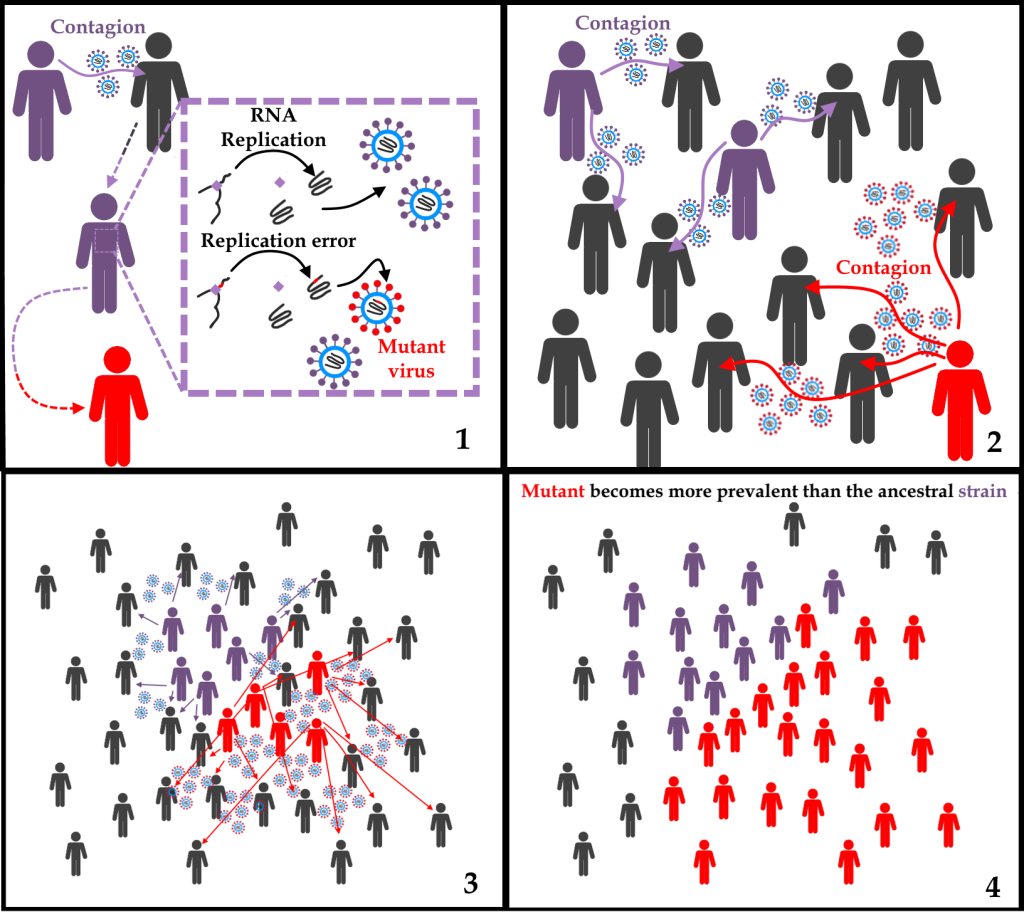 Schematic representing two processes of viral evolution. At the first time point (1), due to random errors to which the RNA replication process is always subject, a mutant sample arises. In the schematic hypothetical example, the change in the genome causes a change in the S protein, which provides increased infectivity of the virus (2). Gradually, the advantage conferred by the higher infectivity (3) makes the virus containing the mutation more and more common. With each new generation of the virus, the natural selection mechanisms continue to act, so that the mutant becomes more prevalent than its ancestor (4) due to the advantage conferred by the mutation.
Schematic representing two processes of viral evolution. At the first time point (1), due to random errors to which the RNA replication process is always subject, a mutant sample arises. In the schematic hypothetical example, the change in the genome causes a change in the S protein, which provides increased infectivity of the virus (2). Gradually, the advantage conferred by the higher infectivity (3) makes the virus containing the mutation more and more common. With each new generation of the virus, the natural selection mechanisms continue to act, so that the mutant becomes more prevalent than its ancestor (4) due to the advantage conferred by the mutation.
Tracking the virus evolution means looking at new lineages and how they differ from one another, what proportion each has in the total species population, and how epidemic trends (growth, stabilization, or increase in the number of cases) are influenced by the distribution of different variants and strains of a virus.
The virus evolution does not necessarily mean that it causes a more severe clinical picture, or that it leads to higher mortality rates. Most of the time, mutations have no significant impact on the virus biology.
Phylogenetics
The study of the evolutionary kinship relationships between organisms – including viral lineages – from the analysis of their genomes is called “phylogenetics”. Phylogenetics is a way of organizing life forms and other biological entities according to characteristics and, prior to the discovery of genetic material and the development of tools for studying it, was carried out by comparing characteristics of organisms, such as structures that have the same origin – for example, human arms and dolphin lateral fins arise from the same structures during embryonic development. More recently, the features being compared are generally genetic, that is, the structure and functionality of genes in common between different biological entities.
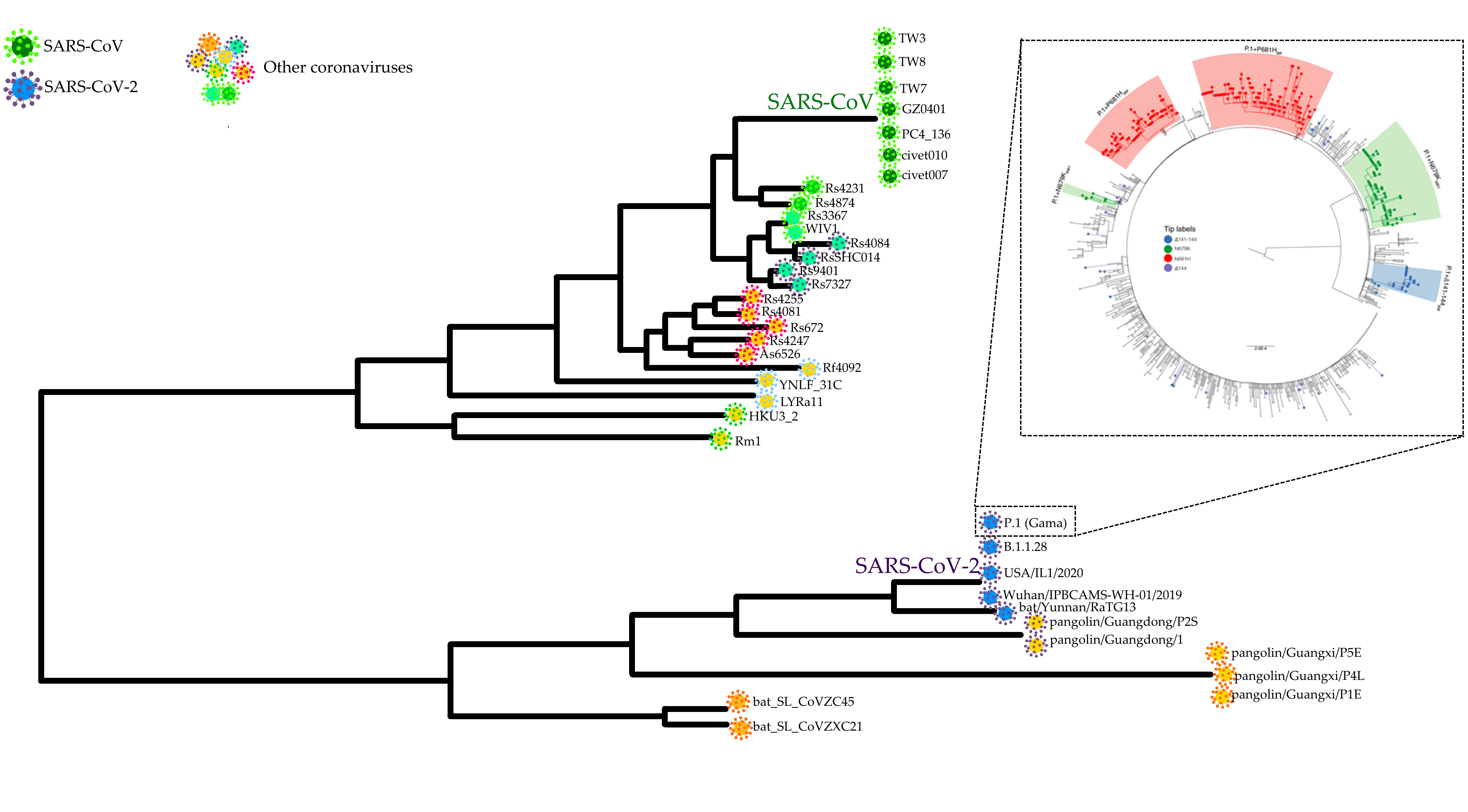
Schematic depicting the kinship relationships between different coronavirus samples, including those causing the SARS epidemic in Asia between 2002 and 2004 and the COVID-19 pandemic. The larger schematic was adapted from Lauxmann et al. (2020), while the schematic detailing the Gamma Variant (P.1) descendants evolution is a reproduction of the schematic prepared by Naveca et al. (2021) for posting on the Virological.Org expert forum.
Immunocompromised
A person who, for whatever reason, has a reduced immune system function. Among the main reasons for impaired immune function are malnutrition, medical treatments such as corticosteroid use or chemotherapy and radiotherapy, diseases such as HIV/AIDS, leukemia, and spinal cord aplasia, and transient situations such as the effects of persistent stress and the immunity suppression naturally associated with pregnancy.
Gene
Genes are portions of the genetic code that, when read and activated (or “expressed”), generate some effect in the cell. The most classic effect of a gene is the manufacture of a protein by an intricate cellular machinery, and the form and function of this protein will be highly dependent on the gene structure that gave rise to it. Genes can also promote or silence the expression of other genes, as well as, in the case of DNA-based genomes, give rise to RNA molecules that will have effective functions within the cell. When, during the genome duplication process, a change in the gene structure occurs, this event is called “mutation”.
Genetics (field of study)
Genetics is the study of heredity. More directly, genetics studies the structures of the genetic material of living things and biological entities such as viruses, with the intent of understanding how the sequences give rise to their characteristics. The study of genetics can be focused on a variety of scales and objectives, from studying a single gene and the relevance of its product (the effect when that gene is expressed, or “activated”) to studying how combinations of genes in a generation can influence the characteristics of its descendant.
Genome
In contrast to genetics, the goal of genomics is the study of genes interacting with each other to form an integrated network of sequences that tend to act in a coordinated way. In its entirety, the genome of an organism or a virus has multiple genes. In organisms with more complex genetics, genes can organize themselves into clusters that are jointly expressed or silenced, as well as sequences that regulate the intensity of expression of other genes. In addition to studying the sequences of individual genes, the study of the genome involves understanding all these interactions between them, and how changes in this balance, due to changes in the environment or mutations in the sequences themselves, can positively or negatively impact organisms.
High Quality Genome (<1% N)
Genomic sequence resulting from precise sequencing, in which there is little uncertainty about the nucleotides at each position in the genome. When there are uncertainties in sequencing, the software associated with the sequencer registers “N” at the position of the ambiguous nucleotide reading. An N percentage of less than 1% means that more than 99% of the genomic sequence is known and the result of reliable sequencing.
GISAID
GISAID (Global Initiative for Sharing Data on Avian Influenza) is an international scientific cooperation initiative created in 2008 to make the efforts to monitor and develop strategies to control circulating influenza virus lineages more collaborative and global.
With databases structure and contact between research institutions involved in viral genomics study, GISAID has expanded the scope of its activities to include the novel coronavirus (SARS-CoV-2) and its lineages in monitoring and cooperation activities. The intent of this expansion is to enable a more efficient and coordinated fight against the spread of the disease by understanding how the epidemic/pandemic develops, as well as near real-time monitoring of virus lineages. The Fiocruz Genomics Network is part of the GISAID international cooperation, and is generating and sharing data about COVID-19 in Brazil.
Spillover Infection
Considered by the authors of a 2017 review paper to be the definitive characteristic of pathogens that cause infection in humans from other vertebrate animal species (infections known as “zoonoses”), spillover is the complex process in which a disease-causing agent in one species or biological group (such as birds or ruminant mammals, etc.) is able to adapt to another biological group of hosts. The process of spreading to other hosts allows the pathogen to cause infections / epidemics in this new species or group of species, and even become endemic in their populations. The same paper argues that for spillover to occur, several factors must be aligned, such as the pathogen dynamics in the original host population, the contact dynamics between humans and the original host (or vectors of the disease), characteristics of the pathogen with regard to its viability outside the host, and individual interaction characteristics of the pathogen with the new host (such as immunity and molecular compatibility).
Lethality
Number of deaths in the affected portion of a population in a given time span. This indicator, unlike Mortality, takes into account only the portion of the population affected by the disease (or risk factor), and not the entire population. Thus, Lethality is a risk measure of a cause for the totality of affected people – in the case of COVID-19, for example, the indicator represents the chance that an infected person will die, while Mortality puts these deaths into perspective in relation to the entire population.
Lineages (virus)
A set of genetically related viruses that descend from a common ancestry. A lineage must have mutations that differentiate it from other variants of the virus. These mutations need not modify any biological characteristics of the virus (e.g., transmissibility or disease-causing potential). To be called a lineage it must have epidemiological relevance, that is, it must be circulating in a large population.
Genetic material
In biology, heredity – that is, the transmission of characteristics from one generation to the next – is based on genetic material. It is a popular custom to say that something is in DNA, but some viruses, such as coronaviruses and HIV, for example, have their genetic material based on RNA. This type of nucleic acid molecule is also present in our cells, but it is involved in cellular metabolism processes and the genetic information flow, acting, for example, in the process of protein production.
The viral genetic material, when inserted into the host cell, is soon read by the cell itself and gives rise to viral proteins that begin the process of redirecting cellular metabolism to produce new viruses. New copies of the viral genetic material are also made based on the material that enters the cells, and it is because of this process of making copies based on the genetic “mold” of the viruses that any mutations can be passed on.
Morbidity
In epidemiology, the concept of morbidity differs from that normally used in the clinic – where the term means the severity of the symptoms of a disease, or how much it debilitates a patient. In the epidemiological context, “morbidity” denotes the proportion of people affected by or carrying a condition, relative to the general population studied, at a given place and time. For example, if in a village of 1000 inhabitants, 100 people are affected by a disease, the morbidity of this disease in the locality is 0.1 or 10%.
Mortality
Number of deaths in a population in a given time span. When dealing with a particular cause (such as COVID-19), one speaks of a cause-specific Mortality. It is important to point out that for this indicator, the total population is taken into account (that is, both people affected by the cause of death in question and those not affected). In this way, cause-specific mortality illustrates the effect of a cause of death on the total population studied, providing a sense of the collective health risk. (For a measure of the effect of a cause of death specifically on the affected population, see lethality)
Mutations
Mutations are errors in the duplication process of the genetic material. Since the DNA (or RNA) that makes up the genome of a biological entity (whether a virus or a living being) is “copied” by enzymes that are subject to errors, small changes can occur in the process, resulting in genomes that are slightly different from the original. Mutation is one of the main mechanisms for generating diversity in biology, since if copies were always exact, no differences would arise between organisms.
These differences are usually neutral or detrimental, but should any change eventually increase the success of the next generation, the tendency is that this mutation will be selected for and become more prevalent with the passage of time. In the case of a virus, for example, a mutation that results in greater efficiency in entering host cells, or in the viral capsid stability – making them able to stay viable for longer in the cellular environment – tends to make the variants that have it more successful than those without the mutation (for a schematic illustration of this process, see Evolution of the Viral Genome above). Mutations that lead to this kind of difference usually directly affect the structure of some gene, the fundamental unit of genetic information.
A mutation can also allow a virus to enter other cell types, and even other species, as in the case of SARS-CoV-2, which probably originated in bats and later developed the ability to cause disease and be transmitted between humans. Other examples are swine and avian influenza, which have a similar history.
Lineage-defining mutations
A mutation or set of mutations that are characteristic of a lineage, verified by genetic sequencing in the samples belonging to that lineage. Lineage-defining mutations can lead to viral genomes with distinct characteristics, such as higher or lower transmissibility, according to the genes affected.
Pandemic
Although the term “Pandemic” has a broad meaning, a 2009 review aimed at understanding the key attributes that make an epidemic event actually considered a “Pandemic” lists the following characteristics:
- “Wide geographic spread” (event without borders);
- “Disease Mobility and Expandability” (i.e., the ability to be competently transmitted by hosts from one locality to another);
- “High attack and transmission rates in outbreaks/irruptions” (multiple cases appearing in a short time span);
- “Reduced (or absent) immunity in the population”;
- “Originality and Evasiveness of the etiologic agent” (not necessarily emergence of a new species, it could be an evolution, a new lineage or serotype, or simply the recrudescence of the same pathogen after a certain period of time when the population is no longer immunologically competent);
- “Infectiousness” (the ability to cause an infection and become established);
- “Contagiousness” (the ability to be transmitted directly from one person to another, with the caveat that pandemics such as the bubonic plague during the Middle Ages had the vectorial route of transmission as the primary route);
- “Symptoms Severity and Lethality” (with high impact on the productive capacity of the population and significant death rates);
It is more than plausible to attribute all the above “Pandemic” characteristics to this one caused by the emergence of the novel human coronavirus, called SARS-CoV-2 and causing Coronavirus Disease 2019, simply called COVID-19. COVID-19 is an injury of great concern, often resulting in severe inflammatory conditions and death. Its likely origin is zoonotic, and its high adaptability to infect the human host, to be transmitted through the air, combined with the fact that it is a new species of virus (originality) to which there has been no previous exposure and therefore no acquired immunity (immunologically virgin population – also called the technical term naïve), contributed to its rapid spread across all continents, facilitated also by the wide mobility of host populations due to a globalized economy and a low preparedness and response level of the surveillance systems of most countries, which had no way of anticipating the new pathogen, nor were they able to act quickly enough.
In technical terms, if a causative agent of Pandemic (or Epidemic) subsequently establishes itself locally in a given population in apparent perennity, possibly causing infection cycles, whether seasonal or sporadic, without total eradication of the etiologic agent, then this pattern is called an “Endemic”.
Infectious virus particles
Viral particles capable of multiplying in a cell and/or tissue, generating an infectious process, with the integral structure, including capsid proteins, phospholipid envelope (when present), surface proteins or glycoproteins – involved in the invasion process of the host cells – and the complete viral genome.
In theory, a single viable viral particle is capable of starting the infection process by finding a compatible host cell, although it is more common that several particles entering a host organism are necessary for the infection process to be successful.
Non-viable particles
Viral particles that, due to the absence of one or more of their constituents, are unable to complete the viral cycle successfully. This inability may be due, for example, to the production of empty capsids (without genetic material), or to the absence of a surface glycoprotein important for the invasion process of host cells, or to defective molecules (either with incomplete or non-viable genetic material, or with proteins or glycoproteins with compromised function due to deleterious mutations).
Endemic Pathogen / Endemic Infection
A disease (or the pathogen causing that disease) can be considered endemic in a locality when it remains there regularly, with low prevalence. Eventually, an endemic disease may cause outbreaks in the region – that is, have an increase in the number of cases – but unlike outbreaks of non-endemic diseases, after a drop in the number of cases the endemic disease remains circulating among
reservoirs and hosts, while maintaining the low prevalence characteristic of an endemic.
Preprint
Initial publication, containing the text and data similar to a finished paper, but published before the peer review process. Because it has not gone through the rigorous process of verification and independent review by other research groups – part of the regular editorial process for publishing scientific papers – the conclusions of a preprint are limited.
This publication model is important for the rapid sharing of information among experts in the midst of a crisis such as the present pandemic, and can inform the media and public opinion as long as the information is treated with extra caution and responsibility, highlighting the fact that the full editorial process is still necessary to gain more confidence in the conclusions presented.
Prevalence
The prevalence of a disease or viral lineage is a measure of the number of people affected by the disease in a given period of time. One lineage is more prevalent than another when, at any given time, more people are sick from it than from the other.
Read Depth
A reliability measure of a gene sequence. The read depth is the number of times a nucleotide has been detected at that position among all the reads in a sequencing process. A high depth means that the error probability (that is, that a nucleotide at a given position is not the one pointed out by the sequencer) is low.
R0
Also called Basic Reproduction Number, in epidemiology R0 is defined as the ability of an etiologic agent to spread, assuming that the population is 100% susceptible to the disease. The number is an estimate of how many people, on average, become infected by coming into contact with an infected patient. Therefore, an R0 of 1 means that each person, over the period of infection, is capable of transmitting the disease to a single person.
When R0 is above 1, the tendency is for the disease to spread so as to cause an outbreak (the number of cases increases, as each patient infects more than one person), while an R0 below 1 means a tendency for the disease to disappear.
Re
While R0 is a measure of the maximum potential spread of an infectious disease, assuming there is no immunity in the population and no measures to contain the contagion, Re is an estimate of the actual number of contagions. Re can vary over time, according to the immunity development (by vaccination or in response to infection), and the adoption of public policies to contain the disease and prophylaxis – measures leading to infection prevention.
In the Covid-19 context, preventive measures, such as social distancing with the adoption of non-presential work, temporary interruption of school activities, and, in more extreme situations, lockdown (a situation in which all non-essential face-to-face activities are suspended and the population movement is restricted to situations of extreme need) are aimed at reducing the Re, i.e., reducing the number of people effectively contaminated by contact with a patient.
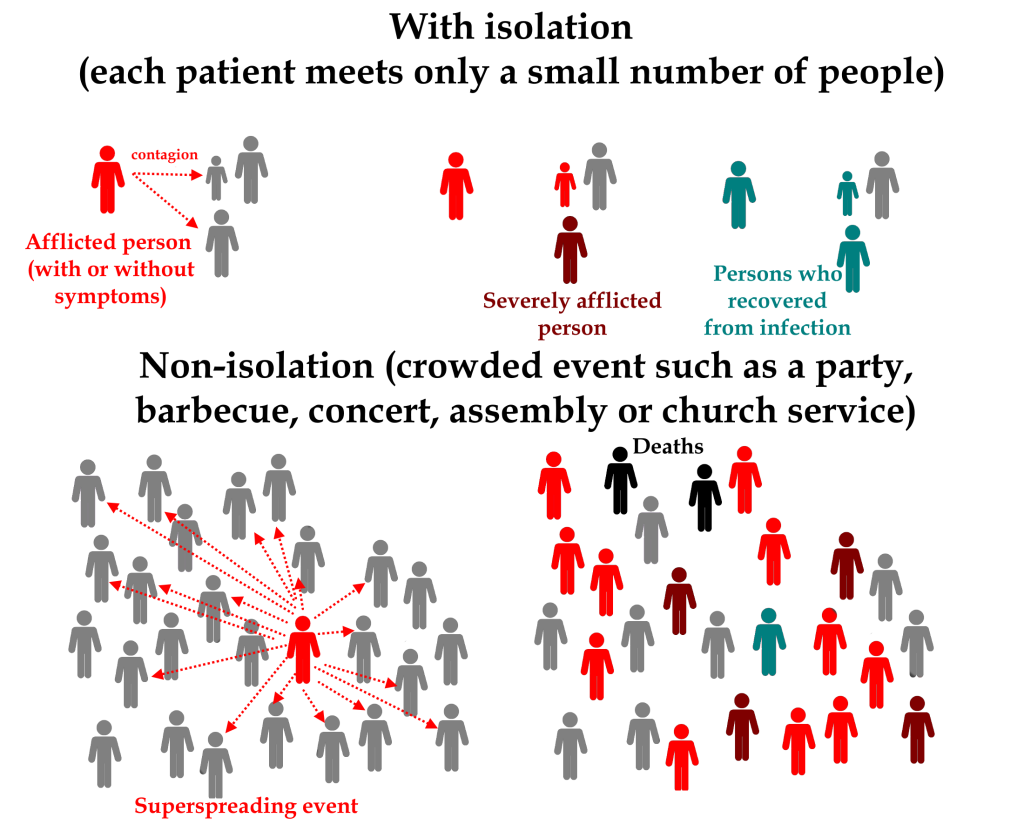 When infectious diseases such as COVID-19 are considered, events in which many people are gathered may result in an increase of the virus’s Re, due to the increased contact between infected and susceptible individuals. Such events are called “Superspreading Events”.
When infectious diseases such as COVID-19 are considered, events in which many people are gathered may result in an increase of the virus’s Re, due to the increased contact between infected and susceptible individuals. Such events are called “Superspreading Events”.
Reservoir
In epidemiology, an animal, a person, or even an environmental factor (e.g., a contaminated water body) is said to be a reservoir for a disease when it maintains the causative agent of this disease in long-term circulation. The infectious agent can be transmitted, directly or indirectly, from reservoirs to hosts of the disease, and can create outbreaks and even pandemics. When dealing with a biological reservoir (person or animal), it is not uncommon for the reservoir itself not to get sick, but only to serve as a “bridge” for disease transmission to susceptible hosts. Reservoirs are also an important factor that hinders the eradication of a disease, because even if the pathogen is completely eliminated among the disease hosts, it will remain circulating among reservoirs.
RT-PCR
This test is used to detect the presence and quantity of RNA in biological samples. Through the use of laboratory synthesized RNA molecules, called “probes” and “primers”, which bind specifically to a genetic sequence of interest – in this case, portions of the SARS-CoV-2 genome – it is possible to detect whether there is viral RNA in the sample and quantify the copy number of the sequence of interest by using fluorescence.
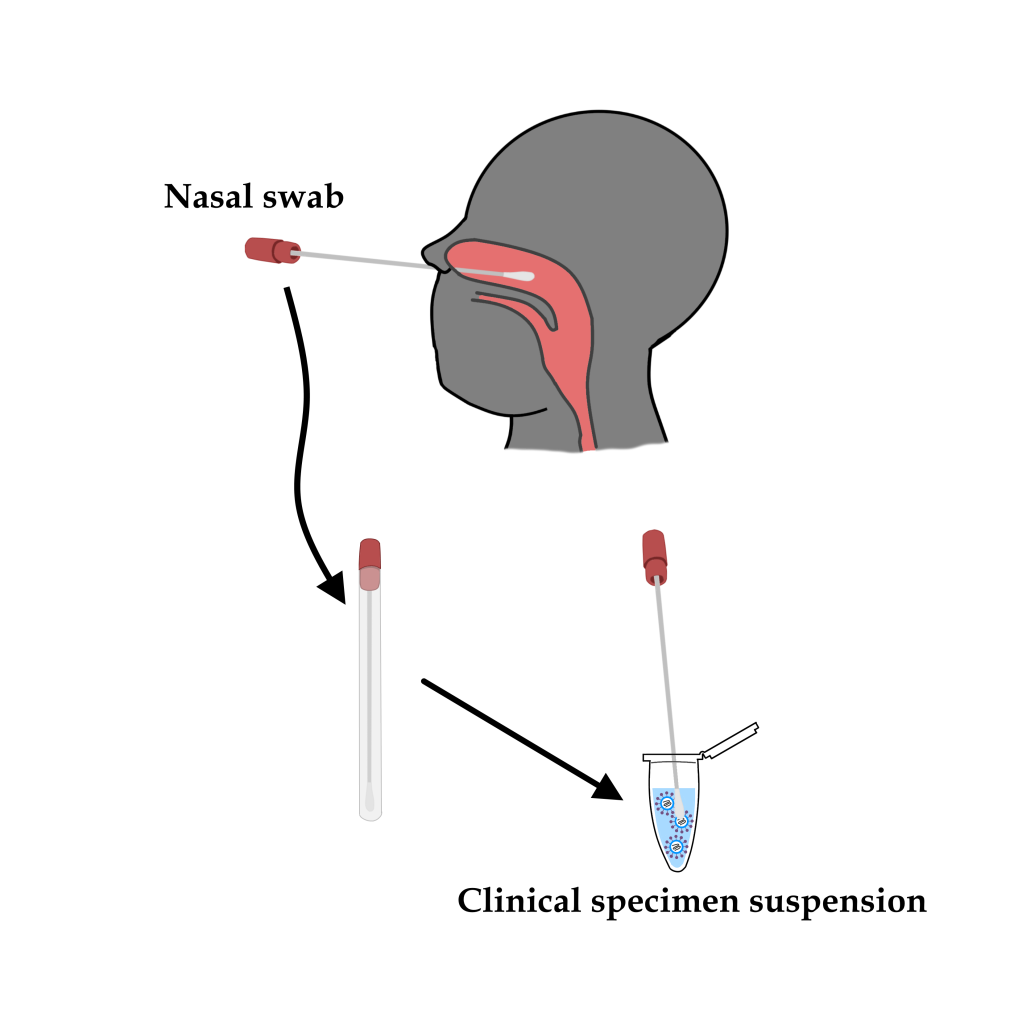
Obtaining a clinical sample for the detection of viral genetic material is done by the nasal swab technique. Using an appropriate swab, healthcare professionals collect mucosal material from the patient’s oropharynx in a suspected case, from which a viral particles suspension is obtained.
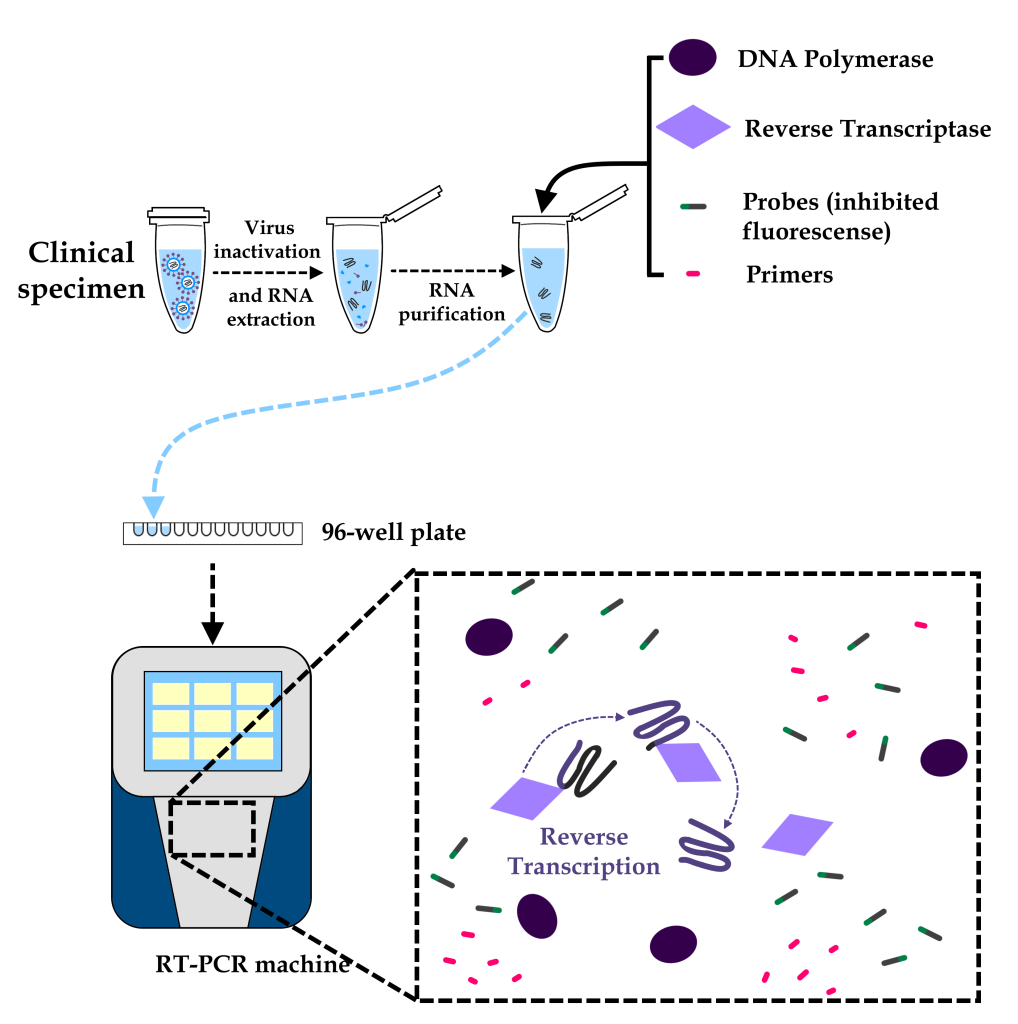
From the clinical sample, it is possible to perform viral RNA extraction and purification. With the purified RNA, the reagents needed to perform the RT-PCR are added to the “mix,” which is distributed on an acrylic plate specifically for this use. In the RT-PCR equipment, on the other hand, the so-called complementary DNA is obtained by the Reverse Transcriptase reaction (RT, which gives the technique its name). It is this complementary DNA – a true viral genetic material copy – that will serve as the basis for further steps.
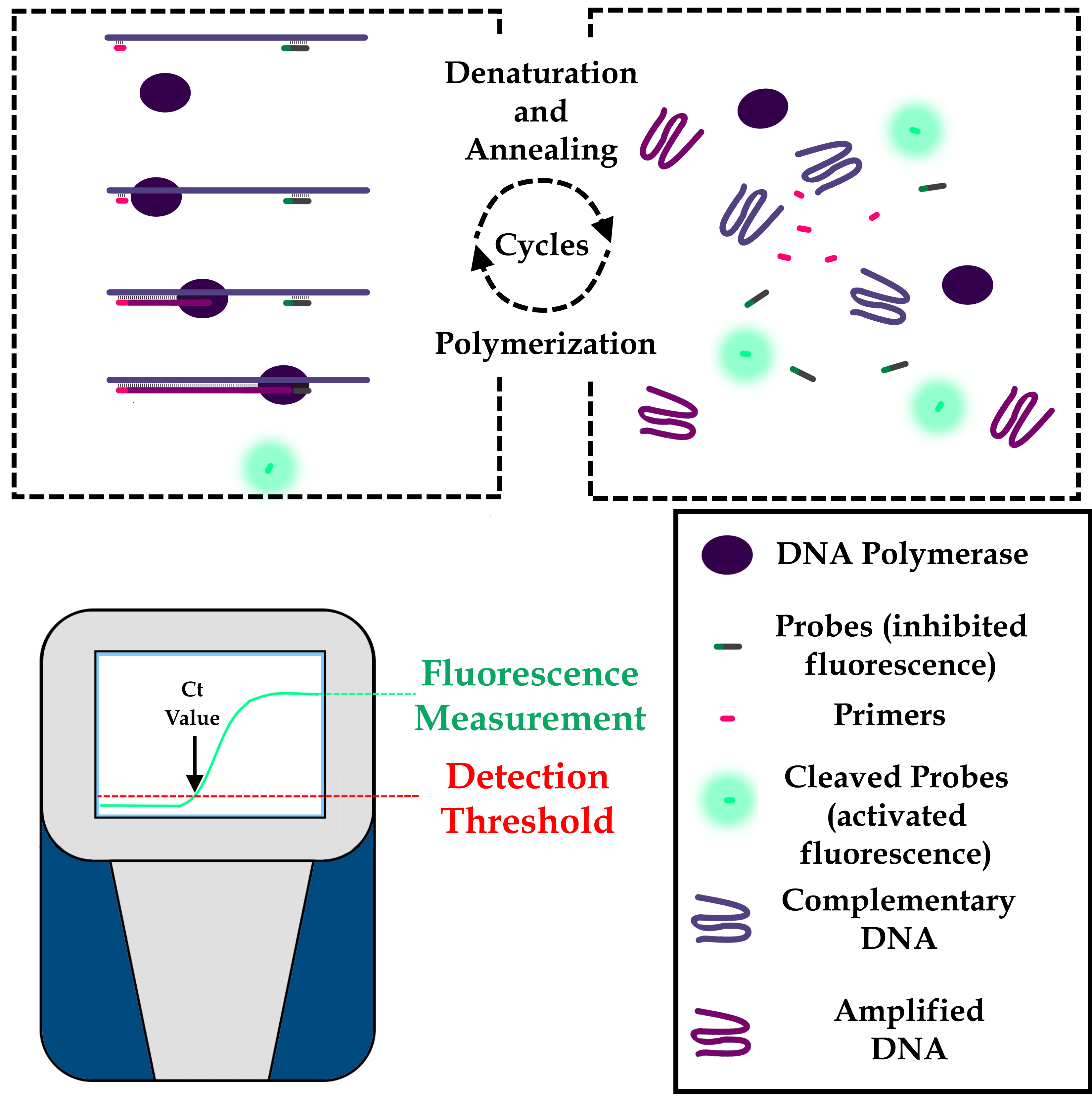
The RT-PCR equipment performs several cycles, which controlled decreases and increases in the temperature of the samples allow the DNA-Polymerase enzymes to make copies of the complementary DNA. By copying the sequences, the enzyme breaks down the probes, which activates the fluorescent tags. Thus, the fluorescence intensity at each cycle is directly proportional to the amount of genetic material amplified. When the intensity of this fluorescence becomes sufficient for the device’s sensors to detect it, the fluorescence threshold is said to have been reached. The number of cycles required for a sample to reach this threshold is called the “Ct value”.
SARS-CoV-2
Severe Acute Respiratory Syndrome CoronaVirus 2. The current pandemic is named because it is the second coronavirus responsible for an epidemic characterized by respiratory syndrome in humans, after the 2003 outbreak in China caused by SARS-CoV. Severe acute respiratory syndrome is a clinical picture that presents a high risk to health and can lead to death.
All lineages and variants of the novel coronavirus belong to the SARS-CoV-2 species.
Epidemiological week
The epidemiological weeks calendar consists of a system to standardize the counting of weeks, to facilitate comparison between different years with regard to epidemiological statistics (for example, comparing mortality from a disease between two or more years). By international convention, epidemiological weeks are counted from Sunday to Saturday, with the first week of the year containing the most days in January and the last week containing the most days in December.
Sequencers
Equipment that makes it possible to unravel the sequence of parts or the whole genome by reading the nucleotide sequences (“blocks” from which DNA or RNA are built). Sequencers are based on the genetic code synthesis reaction, which occurs in a very similar way when cells divide, coupled with technologies to identify which nucleotide is being inserted at a time. Obtaining sequences of genome portions , or of its entire contents, makes it possible to compare different samples, understand how relationships between genes work, and classify samples according to their kinship.
Consensus Sequences
Also known as canonical sequences, consensus sequences, in molecular biology, are the result of comparing several aligned genome sequences. To form a consensus sequence, the most frequent nucleotides at each position are considered. Thus, consensus sequences can serve as a reference for the new samples analysis.
Nomenclature System
A set of rules that must be followed to name a newly found variant of a virus. For SARS-CoV-2, the Fiocruz Genomics Network follows the PANGO nomenclature system proposed by the Center for Genomic Pathogen Surveillance (England).
Seroconversion
Also known as “immune seroconversion,” this is when specific antibodies – produced in response to natural exposure or vaccination against an infectious agent or toxin – can be detected in a patient’s blood plasma. In general, the antibodies production, made by cells called B lymphocytes, starts after a time span of a few days after exposure. In the early stages, IgM-type antibodies are most commonly produced, and later, IgG-type antibodies, so that it is possible to distinguish between recent seroconversion events (presence of IgM and little or no IgG) and older events (characterized by the presence of larger amounts of IgG, and IgM in small or even undetectable concentrations).
SNP
Single Nucleotide Polymorphism is the substitution of a single nucleotide in a gene, when two or more samples are compared. If, for example, in a population, at position X of a gene there can be a Thymine (T) or an Adenine (A), one can say that there is a SNP at this gene position. These differences may influence the evolutionary success of viruses through: changes in proteins that confer adaptive advantages or disadvantages – for example, the different changes in the S protein that allow some SARS-CoV-2 variants to cause reinfection; or an escape from host cell defense mechanisms against invading genetic material (such as interference RNAs).
Transmission Rate
In epidemiological models, the transmission rate is the proportion of individuals in a population classified as “susceptible” who transition to the “infected” state. The transmission rate, therefore, represents the spread of an epidemic through the population, and is different from R0 and Re, although it is directly related to both – for example, distancing measures and vaccination, by reducing Re values, also reduce the transmission rate. Another example is that higher R0 pathogens lead to higher transmission rates, given that the other conditions (such as the intensity and adherence to isolation measures and the susceptibility of the population) are the same.
Ct Value
A PCR reaction (either real-time quantitative or conventional PCR), the equipment performs cycles of temperature change, because each step of genetic material amplification is optimized at a different temperature. In an RT-PCR, you can track how many cycles pass until you have enough genetic material for detection by the device. Since a larger amount of RNA in a sample makes fewer cycles necessary to reach this minimum detection limit, the number of cycles, which is called the Ct-value, is also known as the “cycle threshold”.
Thus, since the Ct-value is inversely proportional to the amount of genetic material analyzed, in the context of SARS-CoV-2 monitoring, different samples can be compared for their Ct-value to assess which have a higher viral load (lower Ct-value). It is important to keep in mind that this is not a direct measure of the number of virus copies in the body of patients, but a consistent indicator that allows for comparative analysis.
Genetic Variants
Any virus that has been sequenced and has mutations that differentiate it from the original version of the virus. The term variant does not imply epidemiological significance and can be broadly used to refer to the genetic diversity of a viral species, i.e., the directory of different versions of its genes. The term strain is also used by the media to denote viral variants, although the use of this term is not a consensus in the virological community.
Some of the most relevant variants and lineages for the Brazilian scenario are:
B.1.1.28
One of the SARS-CoV-2 lineages with likely origin in Brazil, B.1.1.28 was first detected on March 5th, 2020. From this lineage was derived VOC Gamma (P.1), which has caused concern due to its ability to cause reinfection in some patients, as well as higher viral loads.
B.1.1.33
The B.1.1.33 lineage, the main one circulating in Brazil during 2020. It has its origins in cases associated with an ancestral variant – probably from Europe – called B.1.1.33-like. This ancestral strain already possessed one of the two mutations that define the B.1.1.33 lineage (T29148C/I292T, in the nucleocapsid protein), and the second of these mutations is believed to have appeared within weeks of the emergence of the first cases in Brazil in February 2020 (T27299C/I33T, in ORF6). The B.1.1.33 lineage reached a prevalence of over 80% in the state of Rio de Janeiro, while in states like Ceará and Pernambuco its prevalence was lower: 3% and 2%, respectively.
B.1.195
Lineage initially dominant in the state of Amazonas but later bypassed by B.1.1.28 (which was later bypassed by the variant Gama)
Gamma Variant / P.1
Variant of the B.1.1.28 lineage first detected in January 2021 in samples from Japanese tourists who had just returned home after visiting the state of Amazonas. The most likely origin of the variant is in Amazonas itself, around early December 2020. The rapid spread to several regions in Brazil and later to other countries, coupled with the mutations in the lineage, caused Gamma to be classified as a Variant of Concern (VOC). Among the 21 variant lineage-defining mutations, changes in the receptor binding domain (RBD) and the Spike glycoprotein (S protein) result in a protein that is different enough to reduce the ability to neutralize infection by antibodies acquired from previous infections or by vaccination – which may contribute to reinfection. Gamma has 12 mutations in protein S, which include three mutations of concern in common with the Beta lineage (B.1.351): namely, K417N / T, E484K, and N501Y. Preliminary data also point to a possible higher viral load in patients infected with the Gamma variant. If this finding is confirmed, it is possible that the increased transmissibility of this variant is related to the interaction of several factors: the elimination of a higher number of viral particles, a possible increased affinity of the S protein by the receptor present on human cells, and the circulation of Gamma even among the population recovered from previous SARS-CoV-2 infections.
Variant P.2
Variant of the B.1.1.28 lineage first detected in Rio de Janeiro, initially classified as Variant of Interest (VOI) under the name “Zeta”, but currently unclassified as VOC, VOI or VUM. P.2 has 5 lineage-defining mutations, including the S:E484K amino acid substitution, present in variants such as British B.1.1.7+E484K and Gamma (P.1), and apparently associated with the ability to cause reinfection, due to differences in the S protein structure that reduce neutralization by antibodies. Inferences derived from the study of the phylogeny of the lineages circulating in Brazil trace the emergence of P.2 to approximately July 2020. In October of the same year the variant becomes expressive among the samples collected in the state of Rio de Janeiro, becoming dominant in the state by December 2020. As of February 2021, the epidemiological dominance of the P.2 variant is bypassed by the arrival of Gamma in the state, which had a rapid rise in the number of cases.
Variant P.2
Variant initially classified as Variant of Interest (VOI) named “Theta” and first identified in the Philippines; P.3 has some mutations also present in variants of concern, such as E484K and N501Y, and another 5 lineage-defining mutations out of a total of 7. Currently it no longer has an increased risk rating and is neither a VOC nor VOI or VUM.
Variant P.2
Variant not yet classified as VOC or VOI, discovered in May 2021 in the interior of São Paulo in a genomic characterization study of SARS-CoV-2 samples conducted by researchers at the Sao Paulo State University (UNESP). Genetically close to other “P” variants, such as Gamma (P.1), P.2 and P.3, P.4 has among its mutations the L452R substitution, associated with higher infectivity and potential antibody escape.
Delta variant / B.1.617.2
Variant initially classified as VOI, under the name Kappa, B.1.617.1 was initially detected in the Indian subcontinent, but, like other SARS-CoV-2 variants, it soon spread to other countries due to human transit. The Delta Variant has the mutations T19R, (G142D*), 156del, 157del, R158G, L452R, T478K, D614G, P681R, D950N in protein S, with L452R and P681R being mutations of biological relevance. The summation of differences in VOC Delta makes this variant have a higher transmissibility, and possibly escape antibody response, than other VOCs. As the evolutionary process continues to occur in variants, with the selection of samples carrying advantageous mutations, in some localities a variation of Delta, popularly known as “Delta plus”, carrying the mutation of interest K417N, is circulating with greater intensity. Both Delta and its descendants are associated with worsening case and death rates in countries such as India and the United States, so the arrival of this variant in Brazil may pose a collective health risk, considering that complete vaccination coverage (the percentage of the population that has received a complete immunization) is still very low in the country.
Variant B.1.617.1
Variant initially classified as VOI, under the name Kappa, B.1.617.1 was initially detected in the Indian subcontinent, but, like other ..SARS-CoV-2 variants, it soon spread to other countries due to human transit. Variant B.1.617.1 has the mutations (T95I), G142D, E154K, L452R, E484Q, D614G, P681R, Q1071H in protein S. These changes may lead to escape from the antibody response, but this ability is still being studied. At present, the WHO no longer classifies this variant as VOI or VUM.
Mu / B.1.621 Variant
Variant so far classified as VOI. Mu was initially detected in samples from Colombia, but, like other variants of the novel coronavirus, has advanced to other countries – most significantly in Latin America (e.g., Ecuador, 13% of samples analyzed by September 2021; Chile, ~40% of samples by August 2021). The variant has mutations in its genome that are associated with samples that have the ability to be neutralized by antibodies, such as E484K and K417T (also present in the Gamma variant), as well as other genetic changes that are still being studied. The Mu variant has arrived in Brazil, although it has not yet been detected in as significant a volume as other variants such as Gamma and Delta.
Omicron variant / B.1.1.529
Variant of Concern (VOC) first detected in South Africa and Botswana in November 2021. Genome analysis of VOC Omicron samples reveals the highest number of mutations to date among the variants of concern, with about 30 in the Spike protein alone. One such mutation is a deletion in the Spike 69/70 region, associated with failure of PCR amplification of this gene. Such a feature has been used to infer the frequency and monitor the Omicron spread, similarly as was done with VOC Alpha.
VOC Omicron has been shown to have very high transmissibility, and is associated with a new wave of COVID-19 in South Africa, where it has surpassed VOC Delta, and a rapid increase in cases in England. The WHO comments that there is cause for concern due to the presence of mutations such as those in other VOCs that are associated with a further immune escape generated by previous infection with SARS-CoV-2 or by the action of immunizers (vaccination). But it emphasizes that vaccination and infection prevention are the most effective ways to prevent severe cases and hospitalizations.
Virus
Group of non-cellular biological entities. Not having their own metabolism (that is, without organelles and enzymes to carry out chemical reactions, generate or consume energy), viruses are parasites, completely dependent on host cells for their replication – the process of creating new copies, analogous to reproduction. Thus, every virus consists of genetic material surrounded by a protein structure called a capsid.
Some viruses – such as the one that causes COVID-19, SARS-CoV-2 – have a phospholipids layer called the “envelope.” The envelope is similar to the cell envelope, since portions of the host cell membrane are co-opted by these enveloped viruses to form this structure at the end of the replication process. With or without an envelope, every virus needs to have on its surface molecules – usually proteins or glycoproteins – that are used to “stick” to the host cells and place inside them the viral genetic material, which can be based on DNA or RNA, and contains all the information needed to cause infection (including genes for the production of the viral enzymes and proteins by which the parasites master the host cell metabolism and redirect it to produce new viruses)
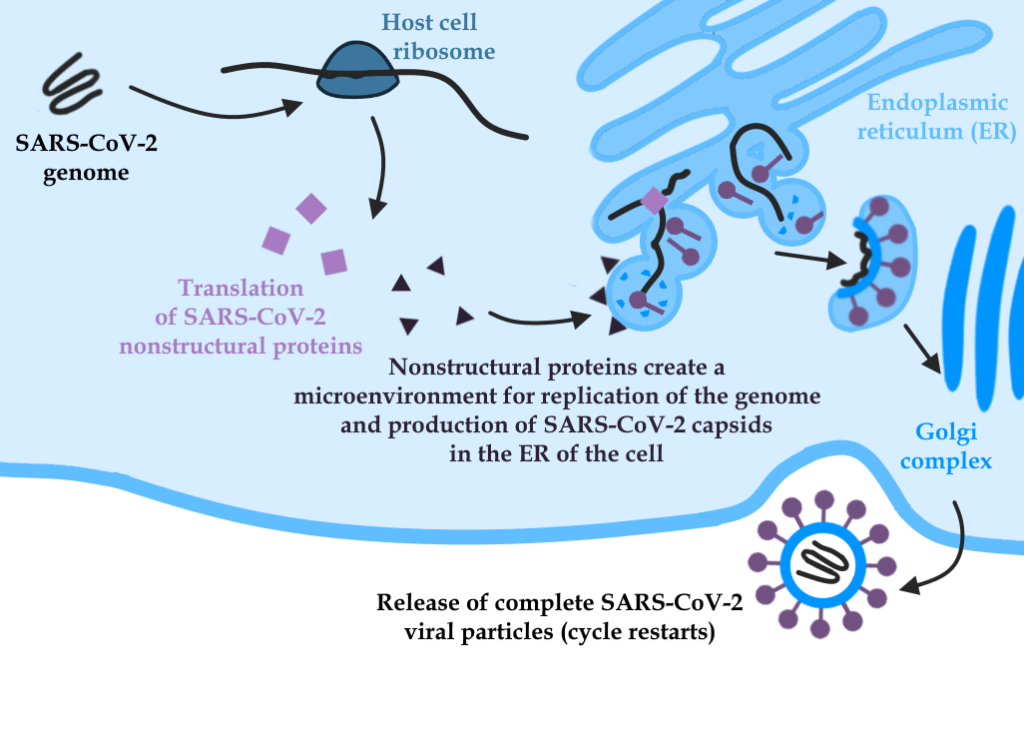
Schematic illustrating the stages of infection subsequent to entry of the viral genome into the host cell. Note that the viral envelope is the result of a mixture of virus proteins and host cell membrane lipids.
VOC
Variants of Concern is a classification given to variants within a lineage when their increased transmissibility (or negative effects on virus epidemiology), virulence (including changes in clinical presentation of the disease), or decreased effectiveness of public health measures (including social distancing and currently available diagnostic, vaccine and therapeutic options at the moment) against the variant in question are demonstrated. Thus, variants such as Gamma (P.1), Alpha (B.1.1.7), and Beta (B.1.351), because of their rapid spread in their countries of origin, the high number of mutations in protein S, and the growing body of evidence for their ability to escape antibodies, have been classified as VOCs.
VOI
Variants of Interest (VOI) is a classification given to mutant gene profiles with the potential to generate a worsening of the epidemic picture, but which have not yet had this potential demonstrated. The classification cut-off line is low – that is, only a few mutations in relevant genes are enough for a sample to receive this classification – so that variants with the potential to cause health situation worsening do not go unnoticed. To be classified as a VOI, a variant must have a mutation that generates amino acid changes possibly associated with changes in transmissibility, virulence (ability to generate severe symptoms), epidemiology, or ability to be recognized by the immune system, in addition to having caused multiple cases of disease/community transmission.
VUM
Variants under Monitoring (VUM) is a classification given to mutant SARS-CoV-2 gene profiles that do not necessarily pose risk, but have characteristics that warrant more cautious monitoring. The World Health Organization classifies lineages as VUMs when some of their genetic changes are suspected to have effects on viral characteristics with potential future risk , but without conclusive evidence about the increased risk associated with these mutations based on current knowledge.